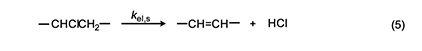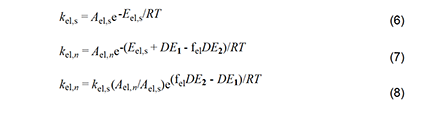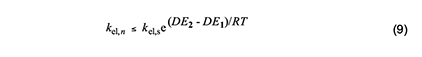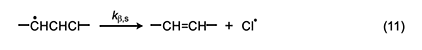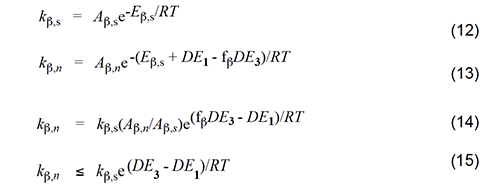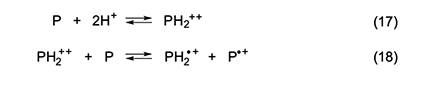|
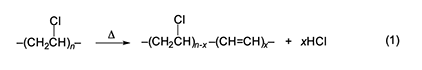
Introduction This paper is a condensed version of the Chemical Pioneer Award address titled “Fifty Years with PVC”, which was delivered in Philadelphia on May 9, 2019, at the annual meeting of the American Institute of Chemists. The round number in the title is slightly misleading, as the author’s work on poly(vinyl chloride) (PVC) actually began in 1973 at AT&T Bell Laboratories (“Bell Labs”). On the other hand, his early ideas for this research were conceived before that time. The intrinsic thermal stability of PVC is much less than that of the other major commercial synthetic polymers. For that reason, an enormous amount of research, both basic and applied, has been done in order to understand the reason(s) for this problem and to solve it. In fact, thousands of papers have been published in this general area. Remarkably, though, a clear understanding of the mechanism of the initial thermolysis process remained elusive for more than 60 years after PVC was first commercialized. The present article outlines the work of the author and his associates relating to this mechanism and then comments briefly on a promising approach to thermolysis prevention. However, no attempt is made here to describe all of the research that was recognized by the Chemical Pioneer Award. A major concern of this paper is the question of how and to what extent free radicals are involved in the earliest stages of degradation, where the polymer loses most or all of its useful properties. Overview, Structural Defects, and Nonradical Degradation Except for the very low concentrations of structural defects it contains, PVC is a conventional head-to-tail polymer made commercially only by free-radical polymerization. In the earliest stage, its thermolysis (equation 1) is just a process of dehydrochlorination involving the sequential loss of HCl from adjacent monomer units, in order to generate a family of conjugated polyene sequences having different lengths. The number of double bonds in these sequences can range from only a few up to 15–20, or even more, depending upon the conditions under which the degradation is occurring. Once initiated, polyene growth is extremely fast, an observation which shows that it involves some sort of “zipper” mechanism in which the loss of an HCl molecule facilitates the loss of HCl from an adjacent monomer unit [1]. The degradation begins slowly at temperatures as low as 100 oC and is autocatalyzed by the evolving HCl [1]. Furthermore, it is now generally agreed to be initiated primarily, if not exclusively, by structural defects that incorporate allylic or tertiary chloride and are situated well away from the ends of the polymer chains [1–3].
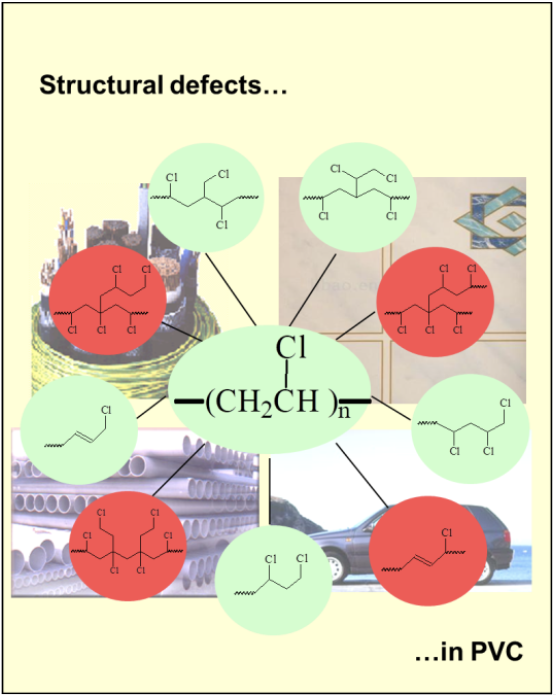
Figure 1 shows all of the defect structures that are now believed to be present in all samples of commercial PVC [2]. The thermally stable structures are shown in green; those activating thermolysis are in red. By using 1H and 13C NMR spectroscopies, together with chemistry involving, inter alia, prior reductive dechlorination of PVC samples with tri-n-butyltin hydride or tri-n-butyltin deuteride, we were able to deduce the molecular constitutions, concentrations (frequencies of occurrence), and mechanisms for the formation of all of these structural defects, while disproving the occurrence of other defects whose presence had been proposed [1,2]. Some of our findings have been confirmed by other researchers [1,2].
Free-Radical Mechanisms Proposed for Polyene Formation Notwithstanding the considerations just described, a huge amount of experimental evidence points to the possible involvement of free radicals in the early stages of the thermolysis of PVC. Described elsewhere in detail [1,2,4–6], this evidence shows that the thermolysis is accelerated by radical sources and retarded (under certain conditions) by radical scavengers, that radicals are produced during the thermolysis process itself, and that conjugated polyenyl diradicals are present in the thermally decomposed polymer.
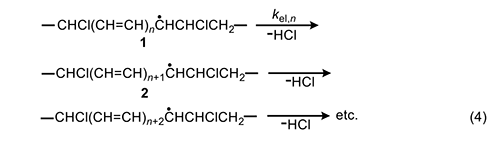
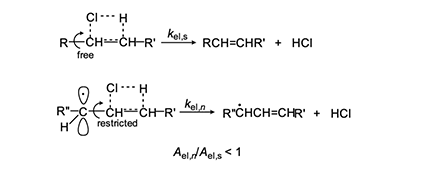
In 1954, Arlman [7] proposed a free-radical mechanism for polyene growth during PVC thermolysis. It is shown in equation 4, where the initial value of n is 0. As depicted, it consists of a series of steps in which HCl molecules are eliminated in succession from adjacent monomer units, so as to form a conjugated polyenyl radical that increases in length as the process proceeds. The rate constant kel,n for each Arlman step may vary as a function of n, thereby giving a set of values that can be compared, as discussed below, with the value of a rate constant kel,s, which applies to a “standard” reaction (equation 5) involving the thermolysis of a simple sec-alkyl chloride containing no radical centers or double bonds.
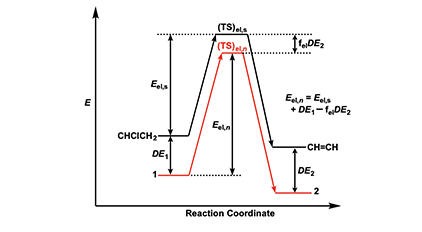
The energetics of the Arlman scheme can be assessed with recourse to Figure 2, where the energy (E) levels assigned to reactant, transition state (TS), and organic product have no quantitative significance, though they are thought to be accurate in a relative sense. The figure shows that the activation energy for the standard elimination is Eel,s and that the ground-state energies of radicals 1 and 2 in equation 4 will be lowered by conjugative stabilization or delocalization (DE1 and DE2), except, of course, in the case where n = 0. To the extent that the transition state, (TS)el,n, for an Arlman elimination (shown in red) structurally resembles 2, its energy will be decreased by some fraction, fel, of DE2. Accordingly, the activation energy, Eel,n, for the Arlman step is given by the expression near the right-hand side of Figure 2.
With these considerations in mind, we can write the Arrhenius equations for the standard elimination (equation 6) and an Arlman step (equation 7) and then combine them to produce equation 8, which could be used with experimental data to predict values of the Arlman rate constant, as a function of n, if the values of fel and the A-factor ratio were known. Since they are not, we will opt instead for a different approach involving the assignment of values to these two terms that are demonstrably too high. The resulting equation can be used to predict maximum values of kel,n, which then can be compared with experimental observations in order to test for the validity of the Arlman hypothesis.
First, we will arbitrarily assign a value of 1 to fel.This assumption is patently ridiculous, as the Arlman transition state certainly would not enjoy all of the stabilization energy of radical 2. Next, as regards the A factors, we note from Figure 3 that, unlike for the standard elimination, rotation about a proximal C-C bond would be restricted, or perhaps even frozen, in the Arlman transition state, because that would tend to decrease the total energy of the system by maximizing overlap of the incipient pi orbital with a p orbital (depicted) or a pi orbital of the radical segment already present.
The concurrent entropic reduction would tend to cause the A-factor ratio to be less than 1. Consequently, assuming it to be 1, together with the assumption that fel = 1, converts equation 8 into equation 9, which can be used with confidence to calculate values of kel,n that are too high. Before proceeding to do this, however, it will be convenient to outline a similar analysis of another free-radical process for polyene growth in PVC.
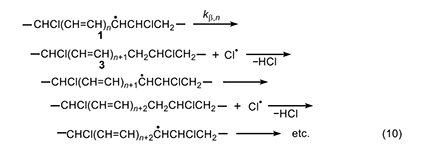
This process (equation 10) was suggested independently by Winkler [8] and by Stromberg et al. [9] in 1959. Like the Arlman mechanism, it starts with radical 1 but requires two steps to form a new double bond and a molecule of HCl. The first step is a β scission process yielding polyene segment 3 and a chlorine atom; the second step involves abstraction, by the chlorine atom, of a weakly bonded allylic methylene hydrogen in 3. This step gives HCl and a new (and longer) polyenyl radical, whose subsequent β cleavage allows the process to continue in the same way. Regardless of which step is rate-determining, if either of them can be shown to be too slow to account for the experimental rate of polyene growth, then the Winkler-Stromberg mechanism can be regarded as disproven.
Accordingly, we will select the β scission step and analyze it by following the procedure used for the Arlman process. In this analysis, the starting species for the standard reaction (equation 11) is now a sec-chloroalkyl radical containing no double bonds, and the Arrhenius expressions for this reaction and the Winkler-Stromberg β scission are equations 12 and 13, respectively. Their combination produces equation 14; and when fβ and the A-factor ratio are assigned a value of 1, this equation reduces into equation 15, which should predict inflated values of kβ,n.
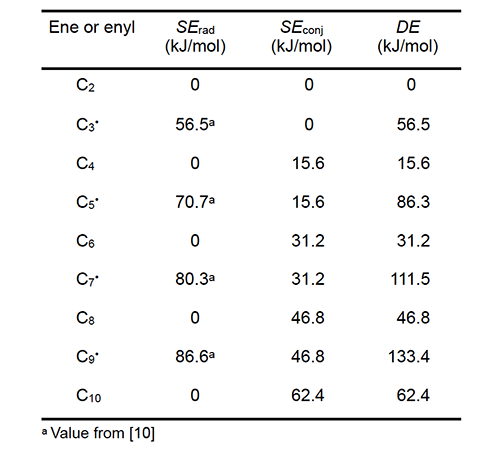
Table 1 lists values of the radical delocalization enthalpy (SErad) and the enthalpy of conjugation (SEconj) for the linear enes and enyl radicals identified in column 1, all of which are conjugated except for C2. For each chemical species, the sum of its two enthalpies is the DE value in column 4. The radical delocalization enthalpies are experimental values from the literature [10]; conjugation enthalpies were determined by multiplying the enthalpy of a single conjugative interaction (15.6 kJ/mol) by the number of these interactions in the species of interest [10,11].
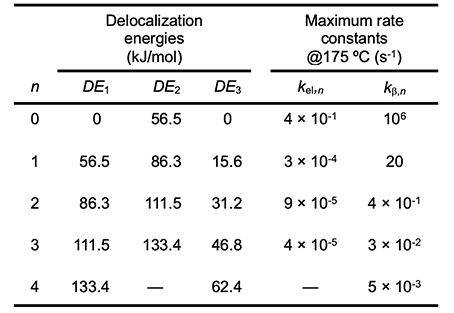
Values of DE corresponding to various values of n are listed in Table 2 for radicals 1 and 2 and for polyene 3 in the schemes of Arlman (equation 4) and Winkler et al. (equation 10). When the appropriate DE1 and DE2 values and the experimental value of ca. 10-7 s-1 for kel,s at 175 oC (references cited in [6]) are used in equation 9, the kel,n values in Table 2 ensue. All of them except the one for n = 0 are much too low to account for the growth of PVC polyenes, which has an experimental rate constant, kp, of ca. 10-2 s-1 at the same temperature (references cited in [6]). Thus the Arlman mechanism can now be dismissed from serious consideration.
At 175 oC, kβ,s is ca. 106 s-1 [12]. Insertion of this value and the requisite values of DE1 and DE3 for 1 and 3, respectively, into equation 15 affords the predicted kβ,n values that appear in Table 2. Though quite high at the outset, they attenuate rapidly with increasing values of n, a trend which shows that the β cleavage step of the Winkler-Stromberg mechanism soon becomes too slow to allow the growth of the longer PVC polyenes at the experimentally measured rate. Other strong arguments against the Winkler-Stromberg hypothesis have been presented elsewhere [6]. Effects of Additives on PVC Thermolysis Rates In order to obtain more information about the mechanism for polyene formation, dehydrochlorination rates of powdered PVC were measured at 180 oC, under preheated flowing argon and in the presence of various additives [5]. β−Carotene (4, nominally all-trans when purchased) was used as a model for long PVC polyene sequences.
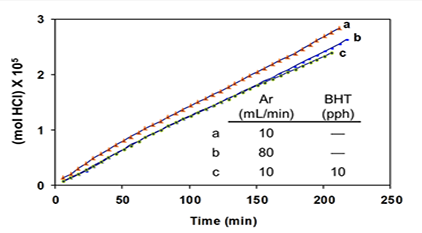
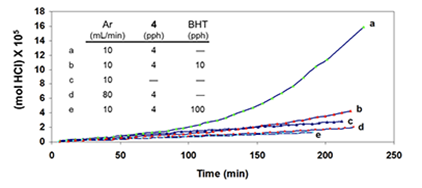
Additives “BHT” (an industrial acronym for 2,6-di-tert-butyl-4-methylphenol) and triphenylmethane (TPM) were introduced as potential radical scavengers (H-atom donors), and metallic mercury (Hgo) was used as a potential specific scavenger of atomic chlorine [5 and references cited therein]. The argon was used to prevent air oxidation and to sweep the evolving HCl into a vessel containing water, where it was titrated with standard base in order to measure reaction rates. Argon also had a third (and very important) function, which was to change the HCl concentration in and around the degrading polymer. The higher the argon flow rate, the lower the concentration of HCl, and vice versa. In the kinetics experiments of Figure 4A, after 200 min, the extent of dehydrochlorination was only ca. 1.5 mol %.
So, at this stage, the polyene concentration was much lower than that. Moreover, the rate (as inferred from the slopes of the plots) was essentially unaffected by a change in HCl concentration (cf. curves a and b) or, when the HCl concentration was relatively high, by the addition of BHT (curve c; pph = parts by weight per hundred parts of polymer).
However, the situation changed dramatically when a small amount of 4 was introduced at the outset. As Figure 4B demonstrates, this additive caused a powerful autoacceleration when the concentration of HCl was high (cf. curves a and c) but not when it was low (curve d), and the autoacceleration was greatly retarded (curve b) or prevented entirely (curve e) by the initial incorporation of increasing amounts of BHT.
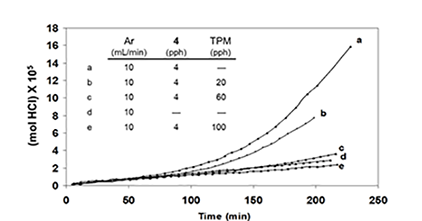
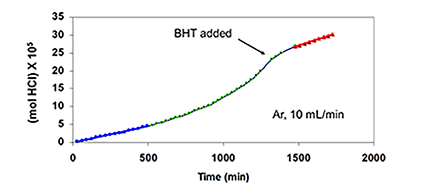
Similar results were obtained when TPM was used as the potential radical scavenger (Figure 4C, cf. curves b, c, and e), although the data showed that, in this respect, TPM was only about one-sixth as effective as BHT. Furthermore, when 4 was eliminated, and the reaction was allowed to run for longer times at high HCl concentration, in order to increase the concentration of polyenes derived exclusively from PVC, rate acceleration began at ca. 500 min and continued until ca. 1400 min, when BHT (100 pph) was introduced (Figure 4D).
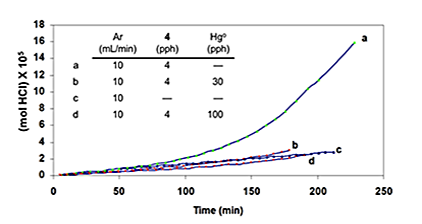
This addition stopped the autoacceleration and reduced the rate to a level that was close to the one observed originally. Finally, as is shown by curves b and d of Figure 4E, when the HCl concentration was high, metallic mercury completely eradicated the autoacceleration caused by 4 and reaffirmed by curves a and c.
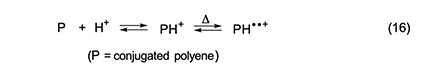
In summary, our study of the rate effects of additives showed that in the thermal dehydrochlorination of PVC, (1) autoacceleration is kinetically dependent upon the concentrations of both HCl and conjugated polyenes; (2) autoacceleration results from a process involving free radicals; and (3) dehydrochlorination does not require free radicals when autoacceleration is not occurring. Mechanism of Autocatalysis During Thermal Dehydrochlorination The kinetically active radical species in PVC dehydrochlorination are conceivably cation diradicals, which, if present, would seem most likely to have resulted from the mechanism shown in equation 16 [1,5 and references cited therein].
Another possibility is that the active species are cation monoradicals formed via the reactions in equations 17 and 18 [5]. Han and Elsenbaumer [13] showed that such a process is apparently a characteristic feature of the protonic acid doping of conjugated polymers, and very strong evidence for the occurrence of its second step was provided by Kispert and his associates [14–17], who used electrochemical oxidation to generate polyenyl dications from a number of carotenoids and verified their ability to oxidize their neutral precursors, as in equation 18. Kispert et al. [18] also showed that 4 and other carotenoids afforded cation monoradicals when they were treated in solution with an excess of HCl.
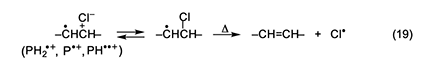
The autoacceleration of PVC dehydrochlorination is most likely to begin with the abstraction, by some radical species, of an unactivated methylene hydrogen, in order to generate radical 1 (n = 0). This radical then initiates a process like that in equation 10 but involving methylene H abstraction mostly from ordinary monomer units. As a result, new isolated allylic chloride structures are formed throughout the polymer matrix, and their subsequent dehydrochlorination in the usual (nonradical) way then can start the growth of new polyene sequences and thus cause autoacceleration. In this overall scheme, radicals obviously play a very important role, but they are not involved at all in the polyene growth reaction. The question that now remains to be answered is the nature of the radical(s) in the very first abstraction step. As noted above, Hgo is a specific trap for chlorine atoms. Therefore, its ability to eliminate autoacceleration completely, rather than merely retard it (Figure 4E), implies that the initial abstraction is performed by atomic chlorine exclusively. This radical species might be formed from any or all of the cation radicals in the system, via equation 19, where P is a conjugated polyene [6].
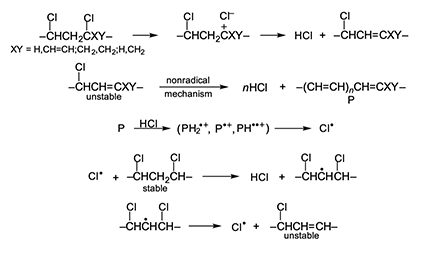
The autocatalysis mechanism proposed here is displayed in full in Figure 5, where the starting structure is either an unstable defect (internal allylic chloride, tertiary chloride) or an unactivated monomer unit, and polyene growth is either ionic (as shown) or quasiionic, depending upon conditions. Post-chlorination studies of PVC have shown that H-atom abstraction by chlorine atoms from the unactivated monomer units occurs mostly from methylene moieties rather than chloromethylene groups [19], as Figure 5 emphasizes. However, abstraction from the latter groups could also lead to the formation of unstable defect structures [6]. Figure 5 rationalizes a huge amount of confusing and contradictory data in the literature, and the explanation for autocatalysis by HCl that it provides is pleasingly consistent with the results of an ozonolysis study of degraded PVC, which showed that the autoacceleration of thermal dehydrochlorination was accompanied by a continual increase in the number of polyene sequences [20].
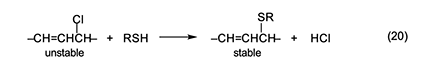
Thermal Stabilization of PVC by Ester Thiols Some of the additives used traditionally as thermal stabilizers for PVC have raised concerns about their effects on human health and the environment. Thus the replacement of these additives has been increasingly desirable. In an approach to this problem, we have studied a class of fully organic stabilizers that are commonly known as “ester thiols”, because they contain at least one SH function and one or more carboxylate ester groups. The following comments relating to these substances are based on a number of publications (e.g., [21,22]) and patents [23–29], which should be consulted for details. Our ester thiols are at least as effective as conventional heat stabilizers in both rigid and plasticized PVC. Some of them also are quite useful as plasticizers when they are introduced at high loading levels. Their excellent performance does not require the presence of metallic or nonmetallic costabilizers. However, in certain cases, their effectiveness is increased by the incorporation of such auxiliaries. When purified, the ester thiols do not have unpleasant odors, and their syntheses are usually rapid, straightforward, and inexpensive. Mechanistic studies with the polymer and with model compounds have shown that a major function of these stabilizers is the deactivation of labile structures by nucleophilic chloride displacement, as exemplified in equation 20. Interestingly, the rate of this reaction increases with increasing RSH acidity rather than RSH nucleophilicity.
This observation suggests that the displacing species are actually thiolate anions, not the thiols themselves, because the acidities of ester thiols, in general, seem much too low to allow these additives to act as electrophilic catalysts for the displacement process. (Thiolate anion concentration increases, of course, with increasing thiol acidity.) The ester thiols also stabilize PVC by scavenging free radicals, via H abstraction from SH groups. Moreover, the radical- or acid-promoted addition of these thiols to the double bonds of polyene chromophores not only reduces color, but may deactivate chloroallyl structural segments that are thermally unstable as well. Our ester thiol patents have been licensed exclusively, for worldwide commercialization, by a company that is incorporated both domestically and overseas. Concluding Remarks The topics addressed in this article have been of major concern to us for some time, but they are not the only ones to have attracted our attention. Omitted here from discussion, for example, are our studies of PVC thermolysis at very high temperatures, new smoke suppressants and fire retardants for PVC (performed, in part, in a collaboration with Prof. R. D. Pike), the polymerization chemistry and microstructures of novel vinyl chloride copolymers, the polymerization chemistry of vinyl acetate, antioxidants and their mechanisms of action, and other areas involving organic and polymer chemistry from a mechanistic point of view. Considerable progress has now been made toward a full understanding of many aspects of the chemistry of PVC. Nevertheless, this fascinating polymer still seems likely to present researchers with chemical challenges (and surprises!) for many years to come. Acknowledgments The author is greatly indebted to his former students, postdoctoral associates, and senior collaborators and colleagues for their invaluable contributions to the research that has now been recognized by the Chemical Pioneer Award. These coworkers are so numerous that their names and affiliations have not been included here. However, many of them are identified in the references. The author also sincerely thanks the following organizations, listed alphabetically, that have supported his research in academia since 1985: Amoco Chemical Co., B. F. Goodrich Co., Colorite Polymers Co., Dow Chemical Co., Edison Polymer Innovation Corp., Egyptian Cultural & Educational Bureau, Elf Aquitaine Inc., GenCorp Foundation, Geon Co., International Copper Assn., Kleerdex Co., National Bureau of Standards, National Science Foundation (Materials Research and Chemistry Divisions), Occidental Chemical Corp., Shell Development Co., Sherwin-Williams Co., Society of Plastics Engineers (four unsolicited grants from the Vinyl Plastics Division), Stockhausen Louisiana Ltd., Stoler Industries, U. S. Department of Defense, U. S. Department of Energy, and Virginia’s Center for Innovative Technology. References
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||