|
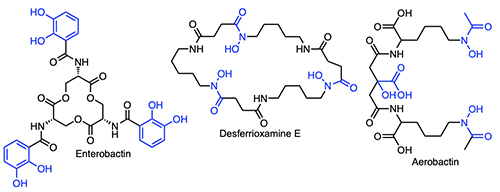
Introduction The majority of bacteria require iron to grow [1,2], yet readily accessible forms of iron are not generally available to microbes because of the low solubility of ferric ion in the environment or because cellular iron is sequestered in proteins and enzymes within host organisms. In contrast to the abundance of iron in the Earth’s crust – being the fourth most abundant element and the most abundant transition metal – the iron concentration in surface ocean waters is vanishingly small – at only 0.01-2 nM across open ocean regimes [3-7]. At the initial point of our investigations, we reasoned that oceanic microbes must either have evolved a special means to sequester iron, despite its low abundance in surface sea water, or that marine microbes make use of metal ions other than iron for key metabolic processes. Many bacteria when stressed for iron produce siderophores – low molecular weight natural products that bind Fe(III) with high affinity. Siderophores can solubilize colloidal iron oxides or in some cases remove Fe(III) bound in proteins, and by so doing facilitate microbial uptake of the Fe(III)-siderophore complex. Siderophores are distinguished by the key functional groups that coordinate Fe(III), including catechols, hydroxamic acids, and α-hydroxycarboxylic acids, among other groups (Figure 1).
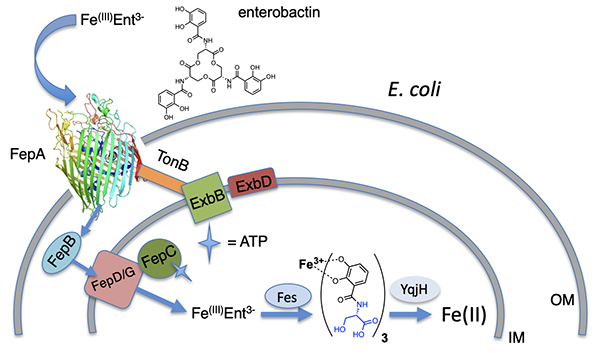
Bacteria generally internalize Fe(III)-siderophores in an active energy-dependent manner tied to ATP hydrolysis (Figure 2) [1,2]. The first point of Fe(III)-siderophore recognition and uptake by Gram-negative bacteria occurs via a specific outer membrane receptor (OMR) protein at the cell surface. After crossing the outer membrane, a periplasmic binding protein and inner membrane transport proteins are involved in transporting the Fe(III)-siderophore to the cytoplasm. Once in the cytoplasm, iron must be removed from the Fe(III)-siderophore complex, which may involve subsequent interactions with esterases or amidases depending on the siderophore, as well as reductases to release Fe(II). The biosynthetic origin, structure and coordination chemistry of siderophores have captured the interests of bioinorganic chemists for decades. Early siderophore investigations demonstrated the staggeringly large proton-independent stability constant of tris catecholate siderophores, such as enterobactin (Ent; 1049) [8] and bacillibactin (BB; 1047.6) [9] for Fe(III). Experiments addressing how iron is released from Fe(III)-Ent3- led to the discovery of the esterase that hydrolyzes each Ser-ester bond in Fe(III)-Ent3- and the reductase that can then reduce Fe(III) to Fe(II) (Figure 2). Even tris hydroxamate siderophores, such as the desferrioxamines, with proton-independent stability constants for Fe(III) at about 1031+1 are still very high [10,11]. Ester hydrolysis may actually occur in the periplasm in some cases. It is a key component for some bacteria such as Campylobacter jejuni, in which the periplasmic binding protein VueE preferentially binds the Fe(III) complex of the enterobactin fragment with two catechol groups and two LSer, i,e, (DHB-LSer)2 [12].
Initial Sortie into the Discovery of Marine Bacterial Siderophores As an inorganic chemist who loves coordination chemistry, I became fascinated by siderophores, which held significance as one of Nature’s biological ligands. As a chemist also fascinated by the marine environment, I was intrigued by the unusual transition metal ion composition of the surface ocean water, with the two most abundant transition metal ions being molybdenum and vanadium, yet iron being quite low, and I wondered about the attendant effect on the bioinorganic chemistry of the marine environment. While bacteria are thought to require mM Fe to grow, Fe levels in the ocean are far less than mM over much of the World’s surface ocean waters. Intriguingly, over 99% of the Fe(III) in surface ocean waters is complexed by a class or classes of organic ligands called “L” [5,7,13]. Many oceanographers asked, “What is L?” While organic complexation of Fe(III) certainly serves to increase the solubility of Fe(III) in the confines of pH 8 ocean water, the origin and identity of these ligand classes is still a topic of much interest. Independent of the nature of L, we became intrigued with identifying characteristics of siderophores produced by open ocean bacteria.
The alterobactins A and B (Figure 3) [14] were the first siderophores we isolated from a marine bacterium. Alterobactin A has an especially high affinity for Fe(III), with a proton-independent stability constant of 1049-53 [14]. The range reflects an estimation of the pKa of each β-hydroxyl proton because it is too high to measure directly. Square wave voltammetry corroborated the high stability constant of Fe(III)-Alterobactin A giving a value of 1051±2 [16]. After characterizing the alterobactins, the 2nd siderophores we identified from an open ocean bacterial isolate was the well-known terrestrial siderophore, aerobactin (Figure 1) [17]. As a result of the discovery of a common siderophore, we very nearly stopped the project, except that we were also working simultaneously on a series of siderophores from other open ocean isolates that had masses varying by 2 or 28 mass units. The close but distinct retention times on the HPLC of these compounds suggested they were related and possibly indicative of desaturation and variation by -CH2-CH22-, respectively, such as could occur in fatty acids. This mass variation turned out to be due to a suite of peptidic siderophores with a series of a fatty acid appendages, as shown for the first two suites of peptidic amphiphilic siderophores we isolated, the marinobactins and aquachelins (Figure 3) [15]. Thus, we didn’t abandon our investigations into marine microbial siderophores.
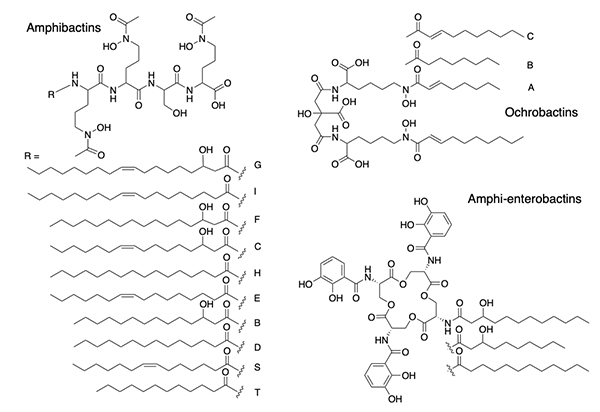
Suites of Amphiphilic Siderophores Suites of siderophores with fatty acid appendages dominated the next set of siderophores we isolated from marine bacteria, with the discovery of the amphibactins (Figure 4) [18], which are rather hydrophobic with only four amino acids in the head group with a wide range in fatty acid appendages [18].
Subsequent discoveries of families of fatty acyl siderophores with variation in the fatty acid included loihichelins [19], moanachelins [20], and pacifibactins [21]. We also discovered citrate derived siderophores, such as the ochrobactins (Figure 4) in which fatty acids replaced the acetyl group of aerobactin (Figure 1), as well as the amphi-enterobactins (Figure 4) which are expanded version of enterobactin (Figure 1). The tri-Ser macrolactone core of enterobactin is expanded to a tetra-Ser core with a series of fatty acids appended to one of the Ser amines in the amphi-enterobactins [22], but they are also sufficiently hydrophobic that they require extraction from the bacterial pellet. The fatty acid may serve as a means for the bacterium to retain its siderophore and to limit diffusion. In general investigations on fatty acyl siderophore partitioning into membranes, we found that siderophores with longer fatty acids partitioned to a much greater extent but also that the Fe(III) complexes of the amphiphilic siderophores partitioned less into membranes than the apo siderophore, which suggests a possible functional significance in the recognition and uptake process for Fe(III)-siderophores [23,24]. The amphibactins have been isolated directly from seawater and identified by mass spectral analysis in comparison to our previous mass spectral characterization [25,26]. Other non-amphiphilic hydroxamate siderophores, such as desferrioxamine G and E (Figure 1) are also reported to be widely distributed in the euphotic zone of the Atlantic Ocean [27].
Photoreactivity of Fe(III) Siderophores with α-Hydroxy Carboxylate Groups The photochemistry of ferric complexes of citric acid is well known [28]. Citrate forms the backbone of several siderophores, including aerobactin (Figure 1), in which the central group is an α-hydroxy carboxylate that is one of the coordinating ligands to Fe(III). In peptidic siderophores, β-hydroxyaspartic acid, is also an α-hydroxy carboxylate that coordinates Fe(III). Thus, we reasoned that Fe(III) complexes of β-hydroxyaspartate and citrate in siderophores would likely be photoreactive and undergo ligand oxidation with reduction of Fe(III) to Fe(II). β-hydroxyaspartate-containing siderophores. The first demonstration of a photoreactive Fe(III) siderophore complex was with the peptidic aquachelin siderophores [29]. Since this time, all investigations of Fe(III) siderophore complexes containing β-hydroxyaspartate have been shown to be photoreactive on UV photolysis into the α-hydroxy-acid-to-Fe(III) charge transfer band. In the case of aquachelin, the photo product retains two hydroxamate ligands (Figure 5) and this binds Fe(III), although with a lower stability constant [29].
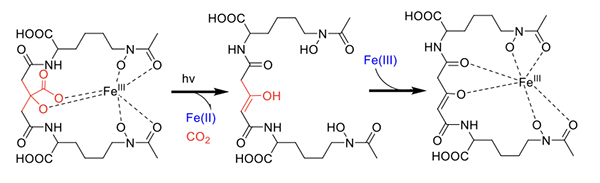
Citrate-containing siderophores. Fe(III)-aerobactin with citrate in the siderophore backbone is also photoreactive (Figure 6) [17], as are the Fe(III)-ochrobactins [24], the Fe(III)-synechobactins [30], and the Fe(III)-petrobactins. In these complexes the reaction is quite clean on UV photolysis into the LMCT band with oxidation of the ligand and reduction of Fe(III) to Fe(II) [31]. The decrease of 46 mass units in the apo-photoproduct reflects loss of CO2 and two H+. The oxidized citryl group in aerobactin produces the corresponding 3-ketoglutaryl derivative, which is in equilibrium with the enoyl derivative used for Fe(III) coordination [31]. We discovered that the affinity of the aerobactin photoproduct is surprisingly high with nearly the same stability constant as for aerobactin [31,32]. UV photolysis of the ferric complexes of the ochrobactins (Figure 4) [24], the synechobactins [30], and the petrobactins [33] produce the same conversion of the citrate backbone to 3-ketoglutarate and coordination of Fe(III) by the enolate form in the photoproduct. As shown above, two structural features dominated the types of siderophores we discovered initially in marine bacteria: that is, a) families of amphiphiles, comprising an iron(III)-binding head-group that is appended by a series of fatty acids; and b) the presence of an α-hydroxycarboxylic acid group in the form of β-hydroxyaspartic acid or citric acid, which are photoreactive when coordinated to Fe(III) [29,31]. Many marine siderophores are both amphiphilic and photoreactive when Fe(III) is coordinated.
Triscatechol Siderophores Form a New Emerging Class of Marine Siderophores Triscatechol siderophores may be another emerging class of marine siderophores in which the catechol is 2,3-dihydroxybenzoic acid (DHB). While the triscatechol siderophores enterobactin [Ent; i.e., (DHB-LSer)3, Figure 1] and bacillibactin [BB, (DHB-Gly-LThr)3], each coordinate Fe(III) with three 2,3-DHB ligands framed on a macrolactone derived from three LSer or LThr residues, respectively, they have not been identified in marine bacteria.
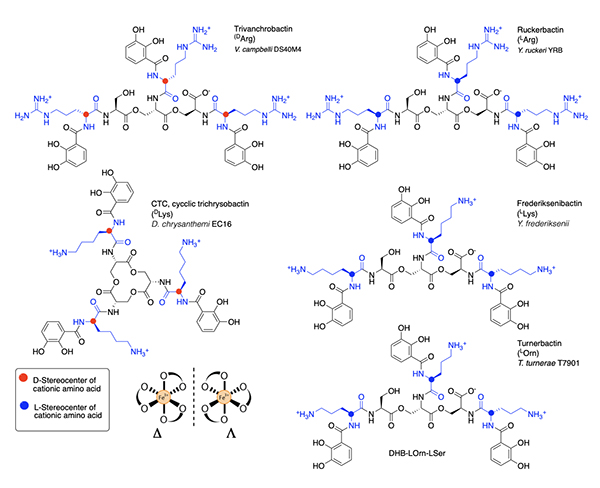
Several related triscatechol siderophores have been isolated from marine bacteria that are distinguished from Ent and BB by the presence of a chiral amino acid inserted between each DHB and the oligo-LSer ester backbone (Figure 7), including the linear tris-LSer scaffolds of trivanchrobactin with DArg (Vibrio campbellii DS40M4) [34], ruckerbactin with LArg (Yersinia ruckerii YRB) [35], and turnerbactin with LOrn (Teredinibacter turnerae T7901) [36]; these bacteria are all marine isolates. The siderophores with LLys and DLys are also known and produced by terrestrial isolates. Cyclic trichrysobactin (CTC) with DLys is produced by the plant pathogen Dickeya chrysanthemi EC16 [37], and frederiksenibactin with LLys is produced by the human pathogen, Y. frederiksenii ATCC 33641) [38]. Trivanchrobactin and ruckerbactin are true diastereomers with DArg and LArg, respectively. Frederiksenibactin with LLys and CTC with DLys are nearly diastereomers, although the former is based on the linear tris-LSer oligoester and the latter is the cyclic tris-LSer macrolactone (Figure 7). The biosynthesis of this suite of D/L cationic amine-containing tris catechol siderophores framed on a tri-LSer oligoester scaffold has been reviewed recently [39], and may well be useful to predicting the full combinatoric suite of this class of siderophores. The stereochemistry of the amino acid inserted between the DHB and the LSer backbone seems to set the stereochemistry of Fe(III) coordination [34,35,37,38]. That is, if an L amino acid is bonded to DHB, the siderophore coordinates Fe(III) in the D enantiomeric configuration, and if a D-amino acid is inserted between DHB and LSer, the L enantiomeric configuration Fe(III)-siderophore confirmation is formed [35,38]. These subtle structural differences suggest the bacteria may have evolved a means to take advantage of the stereochemical differences to promote or control growth.
Summary and Future Directions Our initial foray into marine siderophores started off by screening open ocean water samples for bacteria producing an orange-pink halo when grown on the blue Fe(III)-chromazurol S agar media [40], corresponding to removal of Fe(III) from the blue Fe-CAS complex. We discovered many new siderophores in this manner, although I worried about duplicating our efforts and rediscovering a siderophore we already had previously characterized. The onset of rapid microbial genome sequencing was particularly welcome because it enabled biosynthetic pathways for siderophores to be screened and led to a targeted approach for the discovery of new siderophores. The question of whether oceanic bacteria make use of metal ions other that iron in their metalloenzymes was posed in the introduction. While that question has not been addressed, it is clear that oceanic bacteria are producing siderophores that coordinate and deliver iron to the source bacterium. However, these siderophores may well be serving to sequester and deliver other metal ions to bacteria also. Preliminary work on the alterobactins shows they can complex molybdate and vanadate, although when Fe(III) is added, the other metals are displaced. Yet the catechol siderophores from Azotobacter vinlandii, protochelin and azotochelin have been shown to complex molybdate and vanadate [41], and to deliver Mo(VI) and V(V) to the bacterial cell [42,43]. The uptake of Mo(VI) and V(V) is especially important in nitrogen-fixing bacteria due to the demand for Mo, V and Fe in nitrogenases. Other transition metals form complexes with siderophores, including titanium(IV) which binds hydroxamate ligands in siderophores (e.g., desferrioxamine B) and siderophore analogs [44,45]; manganese(III) which binds to pyoverdine [46], and hydroxamate siderophores [47,48]; copper(II) and zinc(II) which bind to yersiniabactin [49,50]; and even zirconium(IV) which binds to hydroxamate siderophores [51,52,53]. In fact, some siderophores may not actually have a preference for Fe(III) but may be part of a growing class of metallophores [54].
Acknowledgments I am grateful to all the students and postdocs in my research group who have carried out the investigations on siderophores in my lab. Richard Reid was the first brave student to jump in on this project and his efforts led to the discovery of the alterobactins [14]. Much of our work was in collaboration with Margo Haygood, who has been at SIO-UCSD, Oregon Graduate Institute and the University of Utah – she is an outstanding marine biologist and molecular biologist and an especially a tremendous collaborator and friend. I thank the oceanographers and marine chemists who collaborated with us, including George Luther at University of Delaware, Ken Bruland at UC Santa Cruz, as well as Francois Morel who directed the NSF Center for Environmental Bioinorganic Chemistry (CEBIC) from 1998-2008 (NSF CHE 9810248 & CHE-0221978. I am also grateful to current funding from NSF CHE 2108596.
References
|
||||||||||||||||||||||||||||||||