|
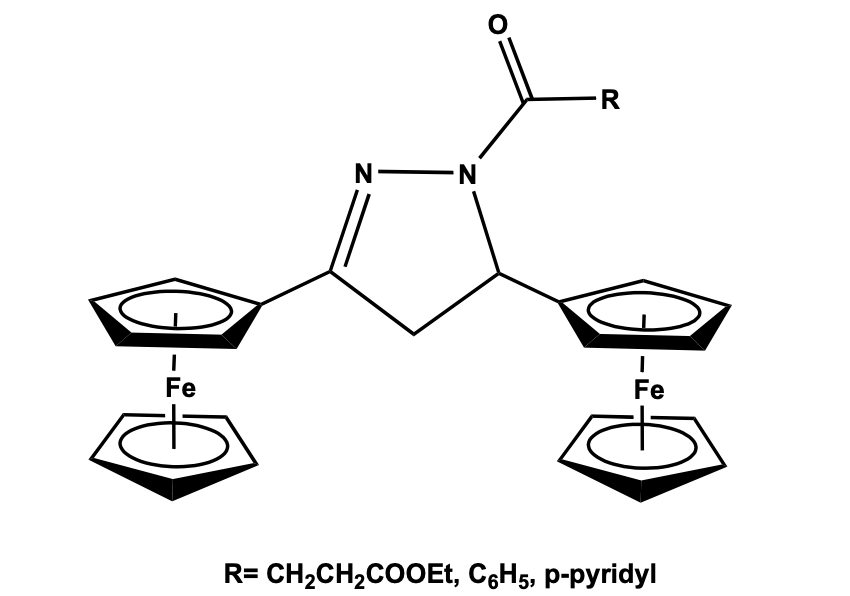
Introduction The synthesis of multi-ferrocenyl molecular systems with specific redox, optoelectronic, magnetic, and conductive properties are important class compounds for several modern technologies [1]. Among the metallocene-based organometallic systems [2], multi-ferrocenyl systems are preferred as potential candidates for molecular electronics due to their intrinsic ferrocene based redox characteristics. Particularly the conjugated and bridging multi-ferrocenyl system with multi-electron redox behavior has great importance because of the formation of mixed-valence species which triggers the migration of electrons within the molecular framework [3]. These switchable arrays have been significantly investigated on the conjugated chain with the ferrocenyl unit for their application in optoelectronic materials. Most of these multi-ferrocenyl systems belong to the mixed-valence system where at least two chemically equivalent ferrocenyl units are electronically coupled and give rise to species with extended electronic communication among the redox units [4]. In this review paper the synthesis, structural feature, properties, and application of multi-ferrocenyl systems will be discussed. Di-ferrocenyl Linkages A variety of di-ferrocenyl compounds have been synthesized in the last decade and their different properties were studied. In recent years, several di-ferrocenyl molecules with conjugated as well as non-conjugated bridging chains have been synthesized to understand their electronic coupling between the redox-active units. In addition to the linearly conjugated bridging ligands between the ferrocenyl moieties, cross-conjugated type bridging ligands are also explored [5-14]. The binuclear and poly-nuclear mixed-valence metal complexes with p conjugated bridging ligand have attracted continuous interest for decades, which led to the study of electron transfer processes. Yuan et al. have reported three dihydro pyrazole bridged ferrocenyl systems by the reaction of diferrocenyl chalcone with hydrazine hydrate followed by acylation with acyl chloride. The diferrocenyl pyrazole derivatives have been structurally characterized by single-crystal X-ray diffraction study which shows that the two ferrocenyl fragments are pointed at two different planes and connected to pyrazole ring carbon atoms. The bond distance between the Cp ring of ferrocene and the Pyrazole group is considerably shorter than the normal C-C bond which confirms distinct conjugation between the ferrocenyl and pyrazole ring (Figure 1) [15].
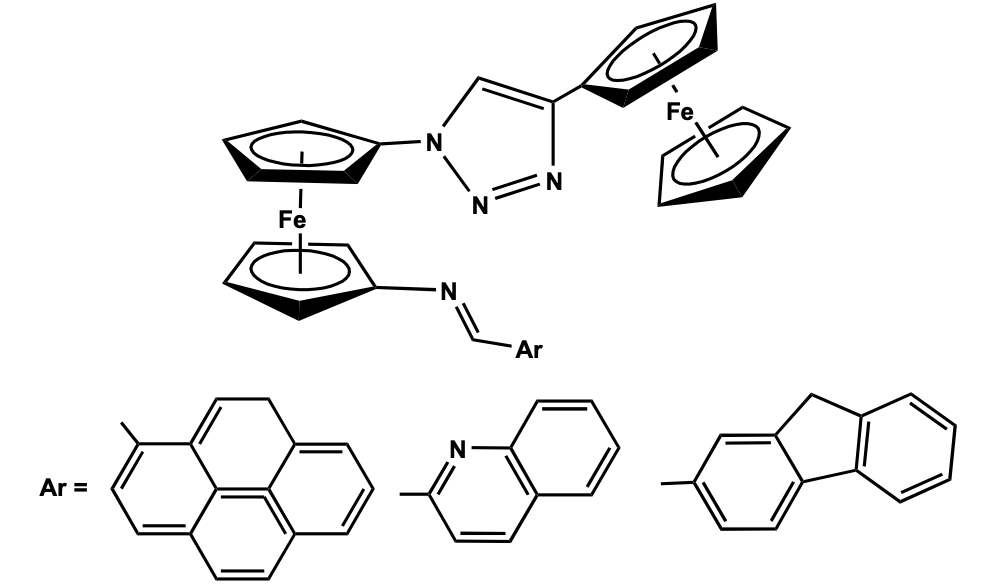
Recently, a research group of Molina has designed a novel synthetic strategy for the preparation of 1,1’- ferrocenyl triazole compound from ferrocene by a series of reactions involving lithiation, click reaction, and Staudinger reaction with trimethyl phosphine to give an iminophosphorane derivative which further undergoes Aza-Witting type of coupling reaction with various aldehydes to give the diferrocenyl triazole derivatives, 1,1’-[Ar-CH=N(h5-C5H4)Fe{(h5-C5H4)(NN=NC=CH)(h5-C5H4)Fe(h5-C5H5)], {Ar= pyrene, quinoline,fluorene} (Figure 2) [16].
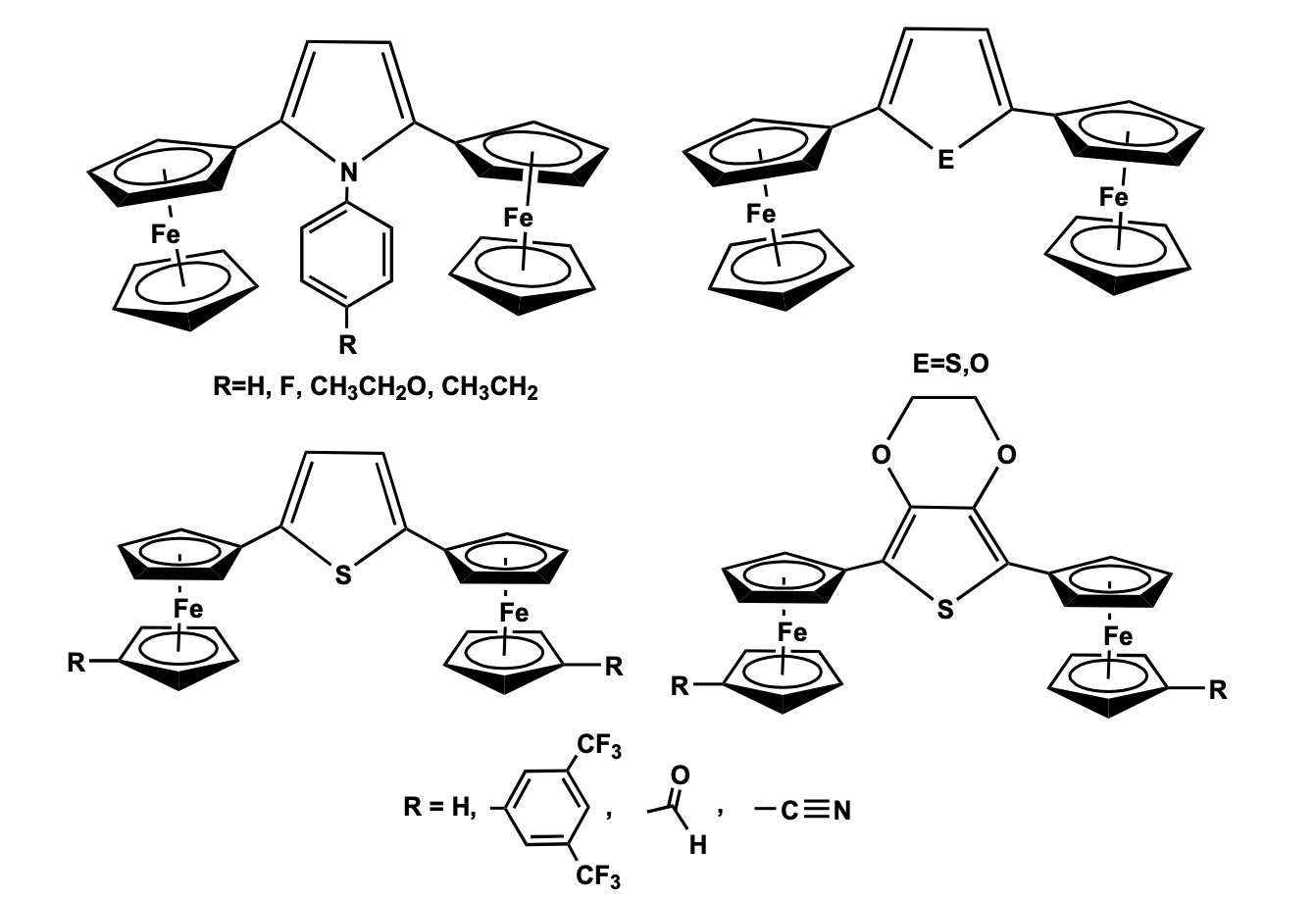
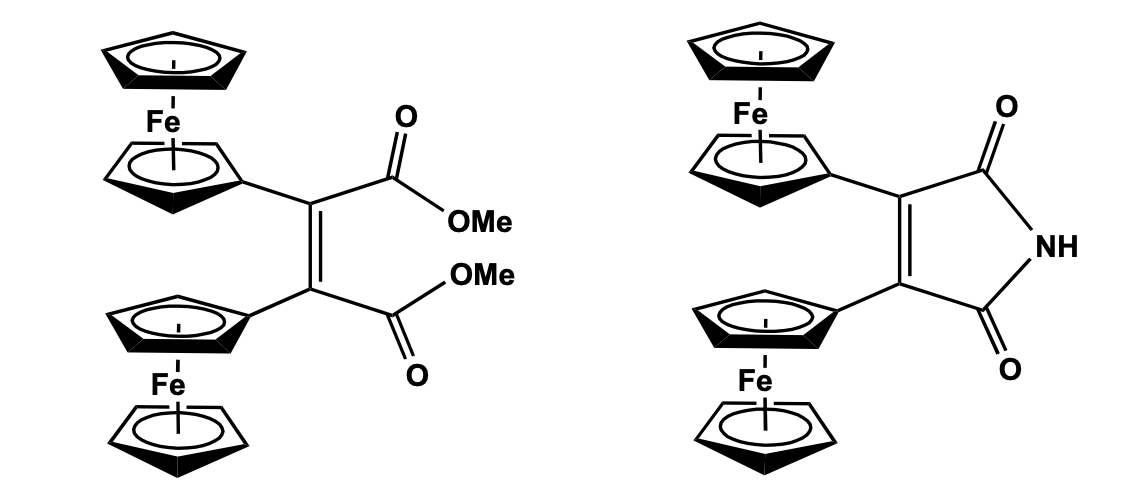
Structural characterization shows that the two cyclopentadienyl rings in the central ferrocenyl unit are arranged in an eclipsed position. One of the substituents on the central ferrocenyl moiety contains a quinoline ring which is coplanar with the imine group and the Cp ring, while the other substituent at the Cp ring is connected via a triazole ring to the terminal ferrocenyl moiety. Cyclic voltammetry study of the compound showed two ferrocene-based redox couples corresponding to the presence of two ferrocenyl units positioned in a different environment. The oxidation potential for the monosubstituted ferrocene has been observed at lower potential as compared to that of the di-substituted ferrocenyl fragment. The compound has been as a receptor molecule showing a distinct shift of the redox potentials due to the addition of Pb+2 and Zn+2 metal cations. The metal recognition properties of the ferrocenyl receptors with Ni2+, Cd2+, Zn2+, and Pb2+ cations have also been confirmed by the change in intensities of the energy bands in the UV-Visible spectroscopy. The binding interaction of the metal ion-receptor system occurs via the imine group of the receptor, the N atom of the quinoline ring, and weak interaction with the triazole unit. Hu et al. has reported a series of 2,5-disubstituted five-membered pyrrole and thiophene bridged di-ferrocenyl compounds and studied their electronic coupling between the redox-active centers. Pyrrole bridged di-ferrocenyl compounds have been synthesized by a one-pot reaction of 1,4-diferrocenyl butadiene with aniline derivative in presence of CuI as a catalyst. 2,5 -Di ferrocenyl thiophene was prepared by the thermal reaction of 1,4-diferrocenylbutadiene with sodium sulfide. Previously, Mathur et al. has reported the synthesis of the thiophene bridged compound using a photolytic reaction of chromium and molybdenum hexacarbonyl in presence of sulphur [17,18]. Recently, the Lang group has synthesized a series of substituted 1 ,1 ’-diferrocenyl thiophene compounds by Negishi C-C cross-coupling method to study the influence of electron-withdrawing substituents at the ferrocenyl moieties and electron-donating function-alities at the thiophene bridge on the elec-tronic behavior of the molecular system [19]. Cyclic voltammetry study of all these thiophene and pyrrole bridged di-ferrocenyl compounds showed two well-resolved reversible redox wave for the two identical ferrocenyl moieties which confirms distinct electronic communication between the two electroactive groups via the heterocyclic bridge. Besides, the influence of the substituent on the extent of redox separation has also been observed and further revealed a class II type valence trap situation in the corresponding mixed valence species (Figure 3) [20].
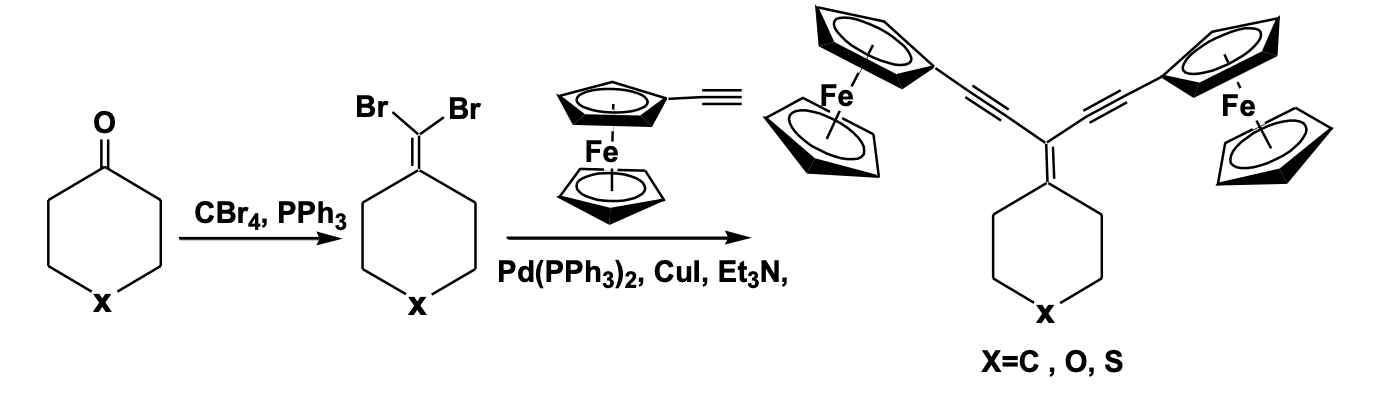
Three geminal-diethynylethene bridged di-ferrocenyl compounds have been recently reported using the Corey-Fuchs olefination process of a keto compound to give a dibromo olefin intermediate, which under Sonogashira condition gave the geminal-diethynylethene bridged di-ferrocenyl system. The structural characterization shows a typical planar Y-shaped geometry of the gem-diethynylethene backbone with the formation of a chair-like conformation of the cyclohexane or thiopyran pendant substituent. The non-bonded Fe...Fe distance between the two ferrocenyl units is in the range 6.76 -9.17 Å, depending upon the bridging substituent. Although two ferro-cenyl units are equivalent, the cyclic volt-ammetric and IVCT study showed a pair of redox couples indicating strong electronic communication between the two redox centers and the presence of a class II mixed valence system within the molecular framework (Scheme 1 ) [21].
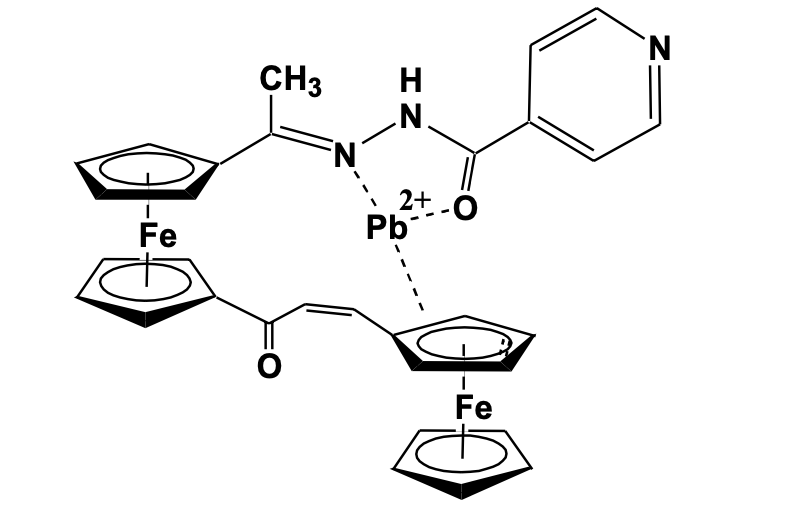
Chatterjee et al. has reported some ferrocene and diferrocene based hydrazone derivatives that can selectively sense Pb2+metal cation. A diferrocenyl based hydrazone–enone compound has shown effective sensing properties. The compound has been synthesized using a two-step reaction method ; the first step being the reaction of 1,1’-diacetyl ferrocene with respective hydrazide to produces the corresponding ferrocenyl hydrazone derivative, which was used for the reaction with monoaldehyde ferrocene in a basic condition to give the diferrocenyl hydrazone-enone derivatives (Scheme 2).
The structures have been confirmed using a single- crystal X- ray diffraction study which shows the presence of two non-equivalent ferrocenyl units oriented opposite to each other and connected to hydrazone and enone moieties. The hydrazone and enone chain in the molecule are oriented in eclipsed conformation. The cyclic voltammetry study shows two reversible one-electron redox waves confirming the presence of two chemically non-equivalent ferrocenyl units. The UV-Visible binding study shows that the compound selectively interacts with the Pb2+ ion with a marked displacement of the spectral bands while other cations show no significant change in their spectral property. Theoretical calculation predicted the site of Pb2+ interaction mainly through the ketonic oxygen, imine nitrogen, and cyclopen-tadienyl ring of the molecular system as shown in Figure 4 [22].
Nemykin et al. has reported di-ferrocenyl compounds (Figure 5 ) with two chemically equivalent ferrocenyl units.
The single-crystal X-ray diffraction analysis shows that the two ferrocenyl units are locked in anti-conformation to each other while the ester and the imide groups are attached to the ferrocenyl unit. The cyclic voltammetric study shows two separate one-electron reversible redox processes indicat-ing substantial electronic communication between the two redox centers. The occurrence of absorption bands in the near-infrared region for the oxidized species confirms intervalence charge transfer and the electronic communication between the redox units [23]. Tri-ferrocenyl Architectures Different types of tri-ferrocenyl systems have been prepared with “star-shaped”, linear, cyclic architecture and their interactions were studied mainly to understand their mixed-valence capacity. The interaction among the ferrocenyl systems occurs through various bridging groups and depends upon the extent of conjugation, chain length, etc. The synthesis and characterization of homoleptic tri(ferrocenyl) “star-shaped” derivatives, with heteroatoms like B, N, P, S, Se, Sb, Bi, has been described by Togni and Hayashi which shows a range of multi-ferrocenyl derivatives with bridging main group atoms [24]. Some early report shows that a tri-ferrocenyl compound with a bridging E-C(O), (E = S, Se, Te) unit was synthesized from lithiated ferrocenyl chalcogenols with ferrocenyl acyl chloride (Figure 6).
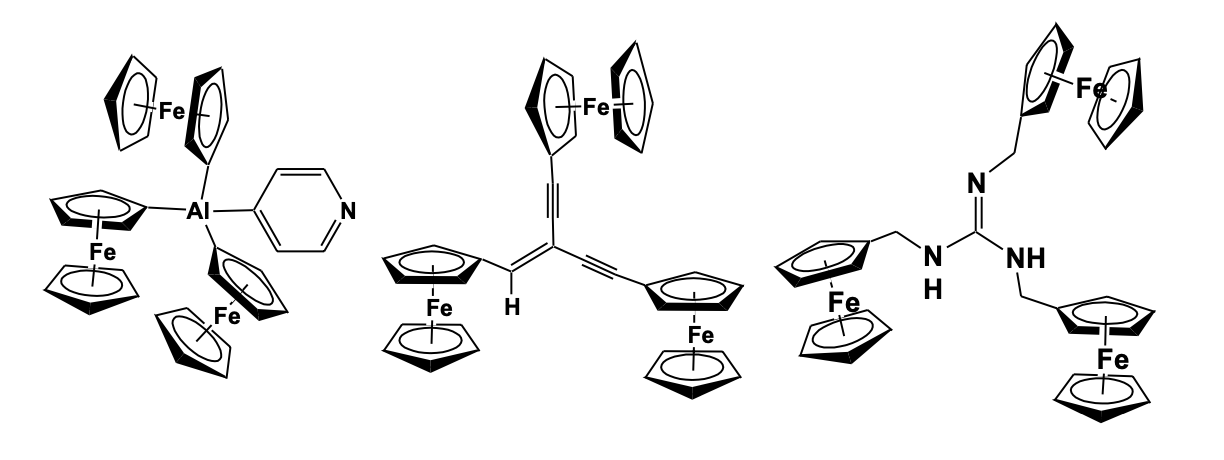
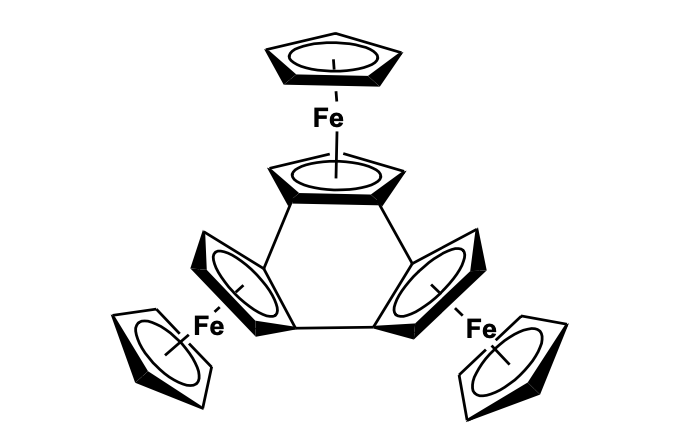
The heteroatom bridged multi-ferrocenyl compounds have shown remarkable chelating properties with various metal fragments [25]. Pena and Cowan described the intervalence electron transfer property of a series of heteroatom-containing tri-ferrocenyl com-pounds, like triferrocenyl boranes, triferro-cenyl phosphine, triferrocenyl phosphine oxide, and triferrocenyl methane, and studied their near IR absorption potential due to the formation of mixed-valence species [26]. Recently, a Si-bridged tri-ferrocenyl compound has been synthesized and their electronic communication among the three ferrocene units was established by cyclic voltammetric study [27]. Wrackmeyer et al. have reported a tri(ferrocenyl)-aluminum-pyridine compound by the reaction of lithiated ferrocene with aluminium chloride and pyridine. The structure shows three ferrocenyl units at different orientations and attached to the Al center, while a pyridine group is also forming an adduct with the Al atom [28]. A tri-ferrocenyl substituted guanidine compound has been synthesized from ferrocenyl azide using a series of reaction steps involving the formation of ferrocenyl isocyanate, a diferrocenylcarbodiimide derivative that reacted with ferrocenyl methylamine. The compound has been found to act as a receptor molecule for the electrochemical sensing of a dihydrogen phosphate anion [29]. Very recently, Low et al. have synthetically designed a conjugated tri-ferrocenyl compound by Pd(PPH3)4/CuI catalyzed cross-coupling reaction of dibromo-ferrocenyl ethane with ferrocenyl acetylene. The compound was characterized by single-crystal X-ray diffraction and the electronic effect among the ferrocenyl moieties on potential electronic communication behavior was studied. Cyclic voltammetry using a strong ion-pair electro-lyte n-Bu4N[PF6] showed broad, over-lapping waves arising from the sequential oxidation of the ferrocenyl moieties in electronically and chemically similar environments (Figure 7) [30].
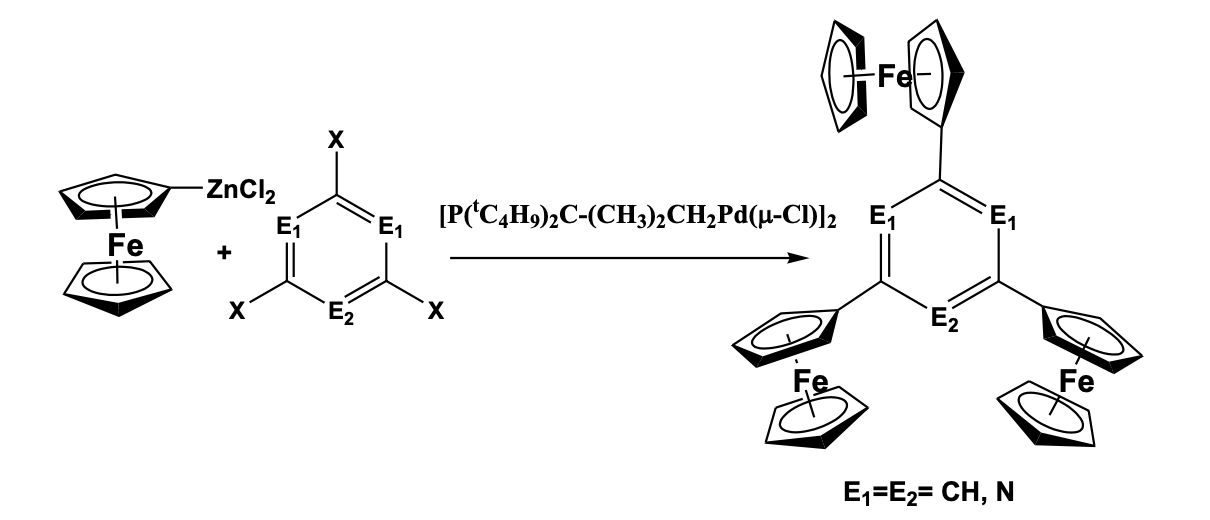
Lang et al. have reported the synthesis of “star-shaped” tri-ferrocenyl based six-membered aromatic ring including benzene, pyridine, and triazine (Scheme 3).
The compounds have been prepared by Negishi coupling reaction between the halogenated aromatic or heteroaromatic ring and ferrocenyl zinc chloride in presence of [P(tC4H9)2C-(CH3)2CH2Pd(μ-Cl)]2 catalysts. Spectral and structural analysis shows that all the three ferrocenes are chemically equivalent while the cyclopentadienyl ligands of three ferrocenyl units are present in eclipsed conformation. The cyclic voltammetry study shows three well-resolved one-electron redox peaks suggesting an electronic coupling across the electroactive groups. IVCT study with the partially oxidized mixed valent complex shows weak intervalence charge transfer absorption due to some electronic communication in the molecule [31]. Appel et al. [32] and Thilagar et al. [33] have separately synthesized redox-active boraxine compounds and established their electronic communication within the redox-active groups (Figure 8).
Single- crystal structural analysis of the selenium analog shows the presence of three ferrocenyl fragments attached to a planar B3X3 ring (X= O, S, Se). The cyclic voltammetry study shows three well-resolved one-electron redox waves indicating sufficient electronic communications between the ferrocenyl centers. The addition of a Lewis base 4-dimethyl amino-pyridine (DMAP) shows the negative shift of redox signals which indicates the poten-tial of these compounds for their use in Lewis acid-catalyzed processes. Synthesis of a tri-ferrocenyl complex of dihydro-1H-trindene has been carried out by a two-step process involving the conversion of dihydro-1H-trindene to bromohydrins by the reaction with N-bromosuccinimide to give tribromo-1H-trindene, which gave the tri-ferrocenyl derivative by Negishi cross-coupling with ferrocenylzinc chloride in presence of PdCl2(PPh3)2 catalyst (Scheme 4).
The electrochemical investigation displayed a single reversible redox wave for three ferrocenyl units in [nBu4N][PF6] supporting electrolyte while two reversible redox wave in [nBu4N][B(C6H5)4] supporting electro-lyte, suggesting influence on the electronic communication behavior by the variation of counter ion during cyclic voltammetry. The intervalence charge transfer study shows near-IR region absorption which indicates the presence of a class II type mixed valence system [34]. Buntz et al. has reported two isomeric tri-ferrocenyl systems containing a fused dehydroannulene and bridged by 1,3-butadiyne linkage (Figure 9 ).
The compounds have been synthesized from 1,2-diethynylferrocene under Hay conditions in the presence of pyridine and CuCl2/CuCl catalyst. The two isomers have been disting-uished by an NMR spectroscopy study which shows two chemically equivalent ferrocenyl units in one isomer while three chemically equivalent ferrocenyl units in the other isomer. Electrochemical investigation of one isomer revealed three one-electron oxidation processes and the other isomer shows two one-electron redox processes indicating the formation of mixed-valence species [35]. Another fused cyclic tri-ferrocenyl compound of triindenyl ligand has been synthesized by an exchange reaction of the potassium salt of trindene trianion with [(h5-C5H5)Fe(h6-fluorenyl)] to give two isomeric compounds, syn-syn-anti and syn- syn-syn (Figure 10).
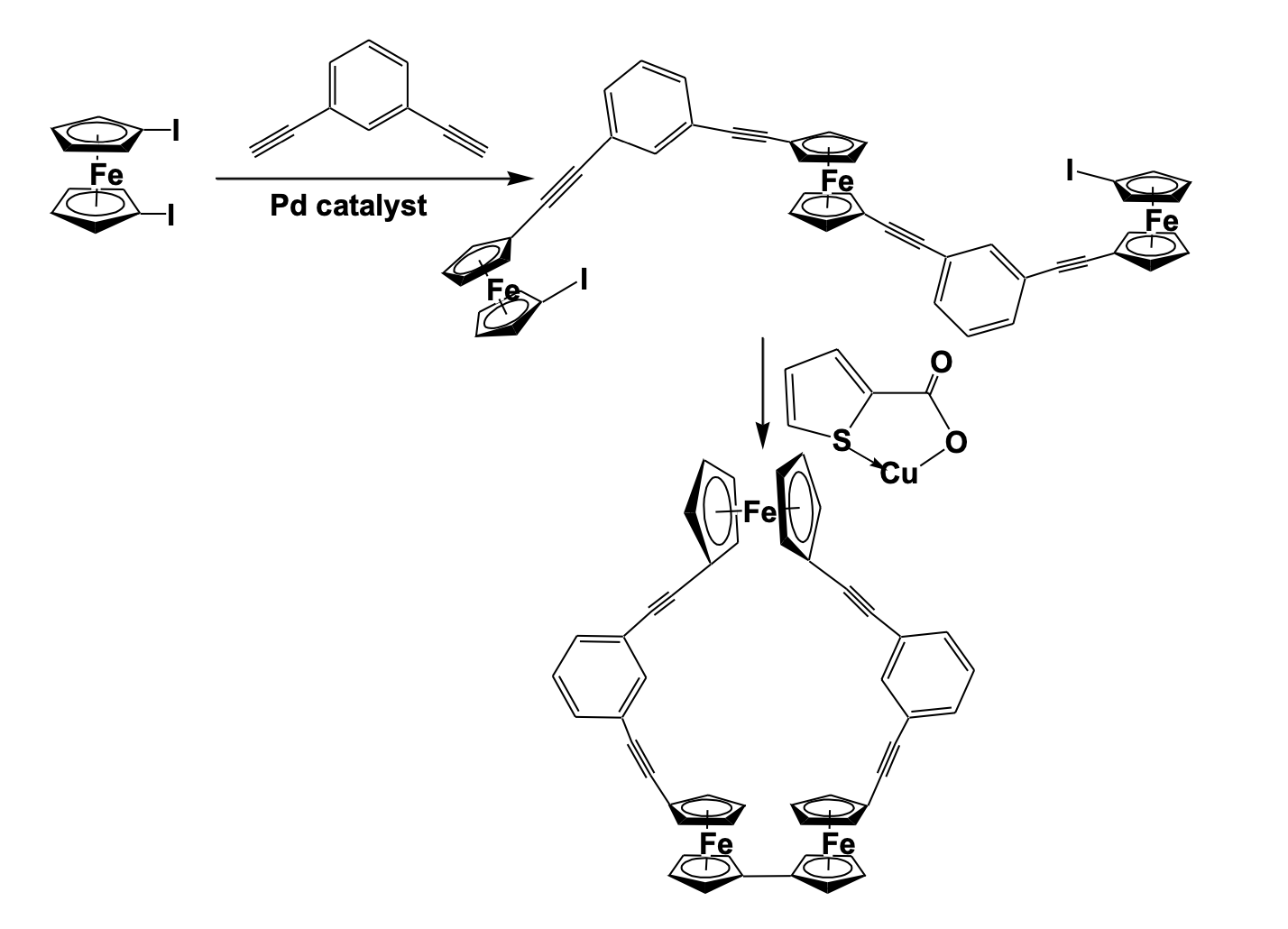
The single- crystal X- ray diffraction study shows the cyclopentadienyl ligand orientation of two syn ferrocenes in staggered conformation while anti-ferrocene in eclipsed conformation. Although the entire system is highly conjugated, the central phenyl ring is not a planer. The deviation from planarity is due to the repulsive interaction between the two syn ferrocene units. The cyclic voltammetric study shows two well-defined reversible redox signals and an irreversible signal while the UV-vis/NIR spectral study shows sufficient electronic interaction among the ferrocenyl centers [36]. Another intervalence charge transfer and the presence of mixed-valence species have been reported by Long using a cyclic ferro-cenyl alkyne compound (Scheme 5).
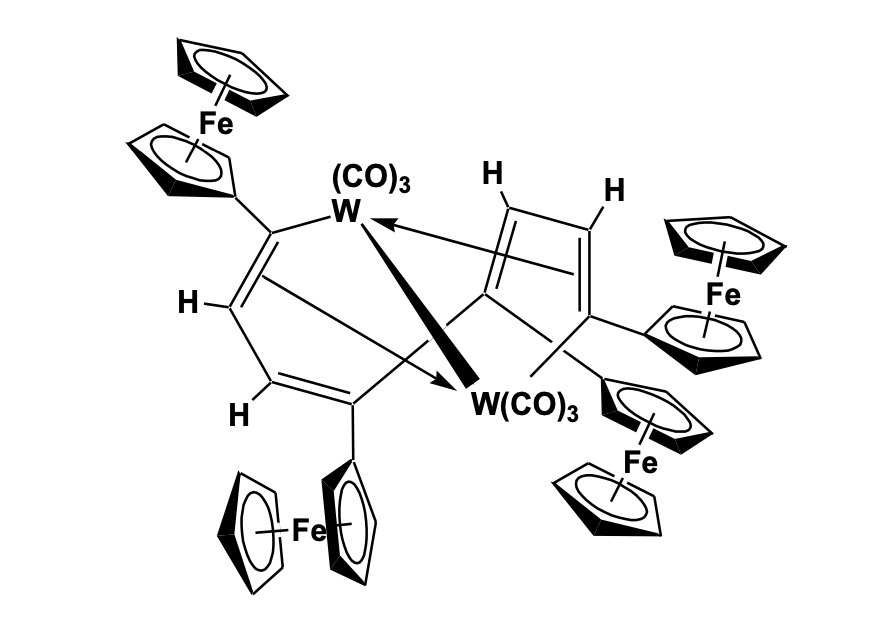
Sonogashira cross- coupling of 1,1’-Diiodoferrocene and 1,3-diethynylbenzene in the presence of Pd(PtBu3)2 catalyst gave a tri-ferrocenyl molecule which was cyclized using a copper-mediated Ullmann-like coup-ling reaction to give a cyclic ferrocenyl structure with alkyne bridging. The structural study confirmed the presence of tri-ferrocenyl macrocyclic molecules with a cavity, which can act as a place for host-guest interaction. The electrochemical interaction between the ferrocenyl units has been investigated by spectroelectrochem-istry. The formation of mixed-valence species has been confirmed by the formation of the absorption band at the near-infrared region [37]. Multi-ferrocene Based Compounds Multinuclear and oligomeric ferrocene complexes have been extensively studied in the last decade for potential applications in multielectron redox catalysis, electron storage devices and as redox switchable molecules. Some of these multi-ferrocenyl systems can selectively vary their electronic properties by oxidation or reduction processes making them important for miniaturized molecular switches and sensor devices. Mathur et al. designed a low-tem-perature photolytic reaction of diferrocenyl diynes with metal carbonyl complexes to give three types of polyyne bridged tetra-ferrocenyl metallacyclic compounds (Figure 11).
Reactions of metal clusters with polyenes have attracted significant interest for the synthesis of extended carbon chains with wide coordination modes. In this reaction, a similar technique has been used to make multi-ferrocenyl linkages which are an attractive candidate for the study of electronic communication and mixed-valence species. The structure of the three tetra-ferrocenyl metalacyclic compounds has been evaluated by single-crystal X-ray crystallography which shows three different cyclic bridges with and without the carbonyl incorporation. The ferrocenyl entities are randomly oriented to release the steric strain across the molecule due to the presence of the bulky -C≡C-CFc group [38]. In another reaction involving ferrocenyl acetylene and tungsten hexacarbonyl, metal-assisted alkyne oligomerization type of rearrangement has been observed to give a unique tetraferrocenyl-cycloocta-tetraene li-
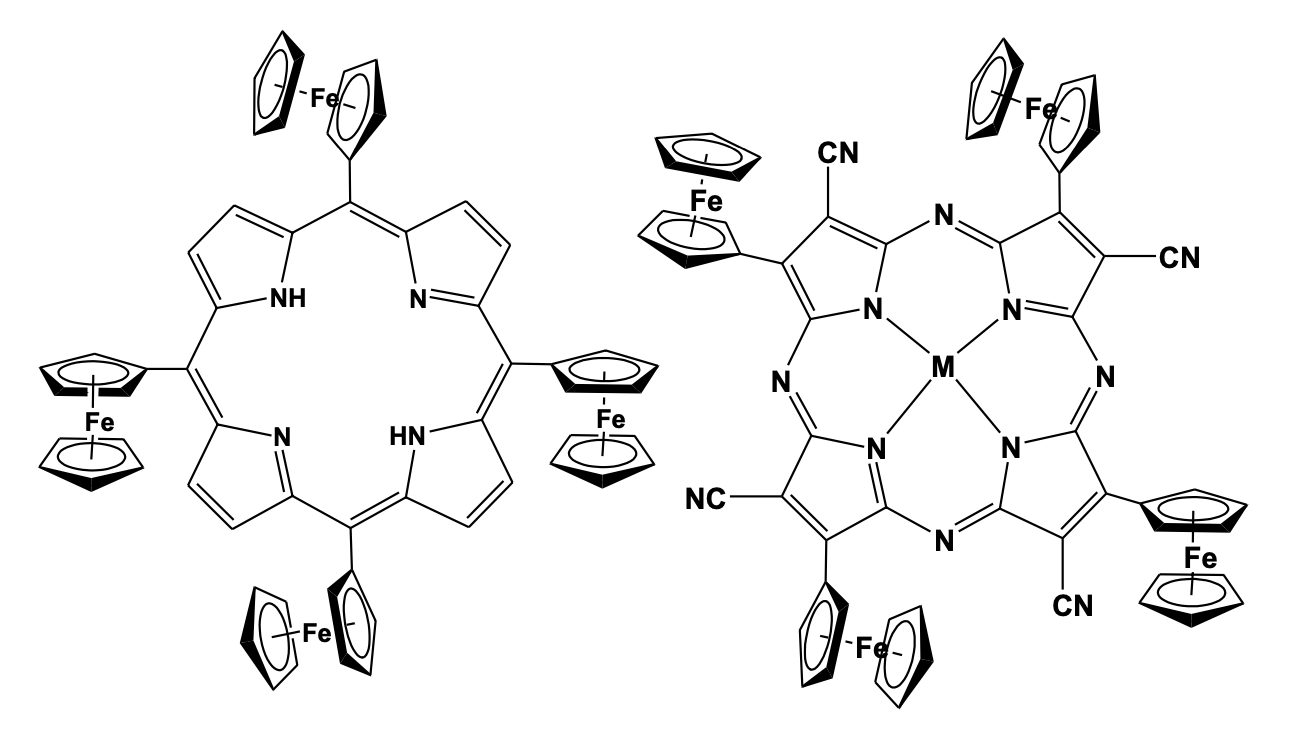
The structure consists of cyclic tetraferrocenyl dimetallacyclo-decatetraene unit pi-bonded to two tungsten atoms, forming a twisted W2C8 ring [39]. Ferrocene linked to porphyrin macrocycle has been an interesting donor-acceptor type molecule with a strong absorbance in the visible spectrum and has been found to show significant electron transfer and redox properties. Porphyrin-ferrocene linkage has shown remarkable optoelectronic properties and is considered to be a potential candidate for their use as molecular electronic devices, in electrocatalysis and sensing of guest molecules. Nemykin et al. has investigated the redox properties of some tetra-ferrocenyl porphyrin type molecule and observed intervalence charge transfer bands in the near-IR region. The charge transfer analysis confirmed the presence of class II type intervalence charge transfer characteristics involving ferrous/ferric centers [40,41]. Synthesis of the metal (M) complexes of tetra- ferrocenyl porphyrin with Zn, Ni, Co and Cu, and their structural study, shows a strong influence of the central metal atom on the degree of non-planarity of the porphyrin ring system which also confirms the presence of numerous metal-to-ligand charge-transfer bands coupled via a configurational interaction, with expected intra-ligand p–p* transitions [42]. Galloni and Nemykin further synthesized tetra-ferrocenyl porphyrin based self-assembled monolayers and investigated their photocurrent generation efficiency [43]. The data of the monofunctionilized monolayers revealed their potential as candidates for the construction of molecular electronic devices. Some of the tetra- ferrocenyl porphyrin derivatives have been used as electrode modifying layer in combination with Prussian blue due to its unique electronic properties and synergic effect between porphyrin ring and peripheral ferrocenyl group. The conjugate has been tested for the sensing of dopamine showing a satisfactory analytical response comparable to other chemically modified electrodes. A magnesium complexed tetraferrocenyl tetraazoporphyrin has been investigated for its use as an energy acceptor from CdS quantum dots as donors. The study showed that CdSe quantum dots can be promising energy transfer donors for NIR-absorbing tetraferrocenyl tetraazaporphyrins to form antenna systems with enhanced light-harvesting efficiency (Figure 1.24).116,117 (Figure 13 ) [44].
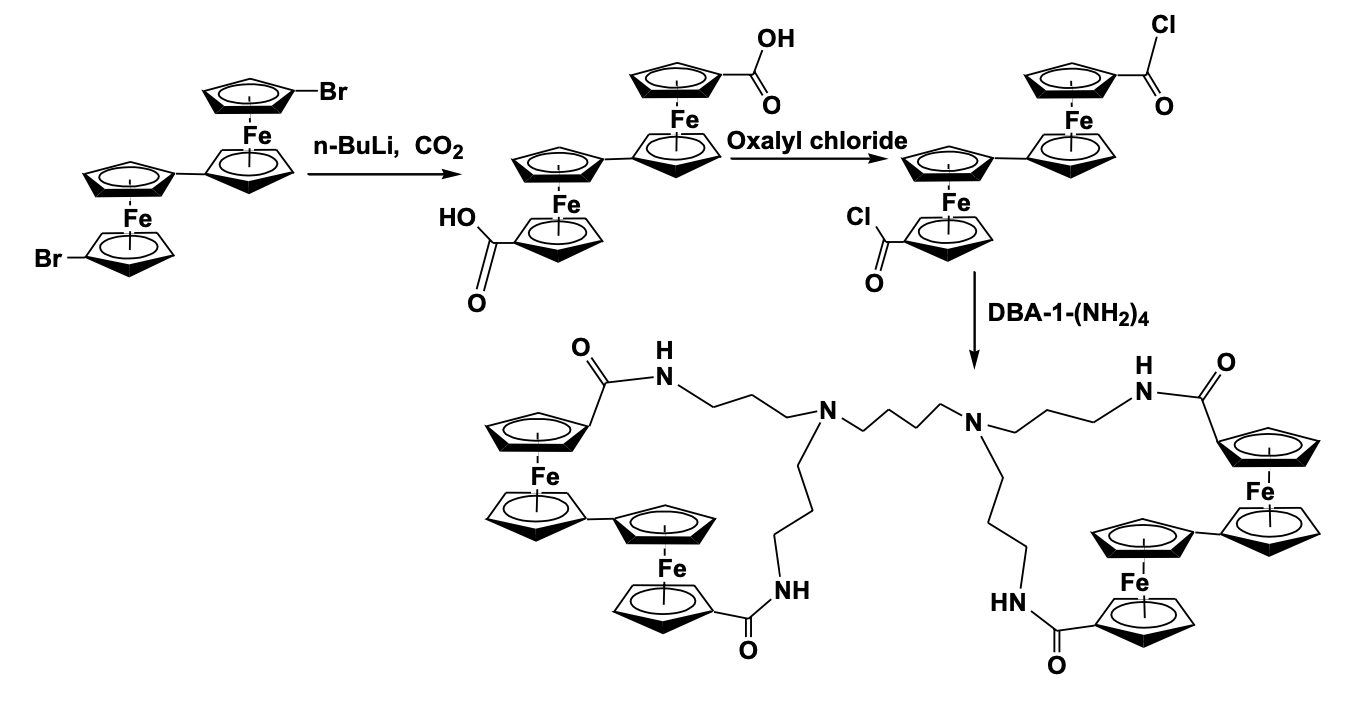
A diaminobutane poly(propylene imine) dendrimer functionalized with biferrocenyl units has been synthesized which shows electrochemical anion sensing properties [45]. The compound, synthesized by the reaction of dichlorocarbonyl bisdiferrocenyl derivative with diaminobutane-based poly(propylene imine) dendrimer, shows two reversible redox waves in the cyclic voltammetry study and shows a large cathodic shift due to the interaction of dihydrogen phosphate and hydrogensulfate anion (Scheme 6).
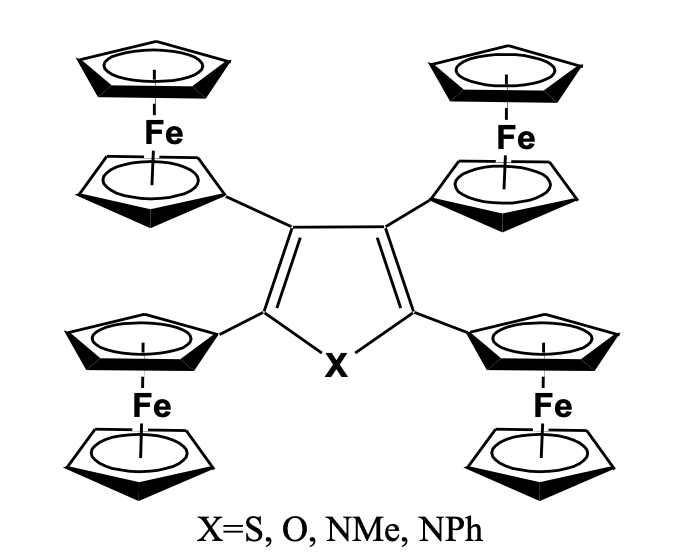
A range of tetra-ferrocenyl heterocyclic derivatives has been synthesized for their study in understanding electronic commu-nication across the heterocyclic moiety. The iron-iron distances in these tetra-ferrocenyl heterocyclic systems have been kept con-stant while the heteroatom was changed to S, O, NPh, or NMe groups. The cyclic voltammetry study shows two reversible redox processes in all the system indicating effective electronic communication between the ferrocenyl units and shows the formation of mixed valence species, confirmed by the appearance of inter-valence charge transfer band at the near-infrared region (Figure 14) [46-48].
Higher analogs of multi-nuclear ferrocenyl systems are not only difficult to synthesize but also pose challenges to stabilizing the molecular system. To find new synthetic methods to obtain higher ferrocenyl compounds with a conjugated network, Peris et al. have designed a series of palladium-catalyzed reactions directed towards the synthesis of a penta-ferrocenyl complex with a conjugated link in between the ferrocenyl moieties (Figure 15).
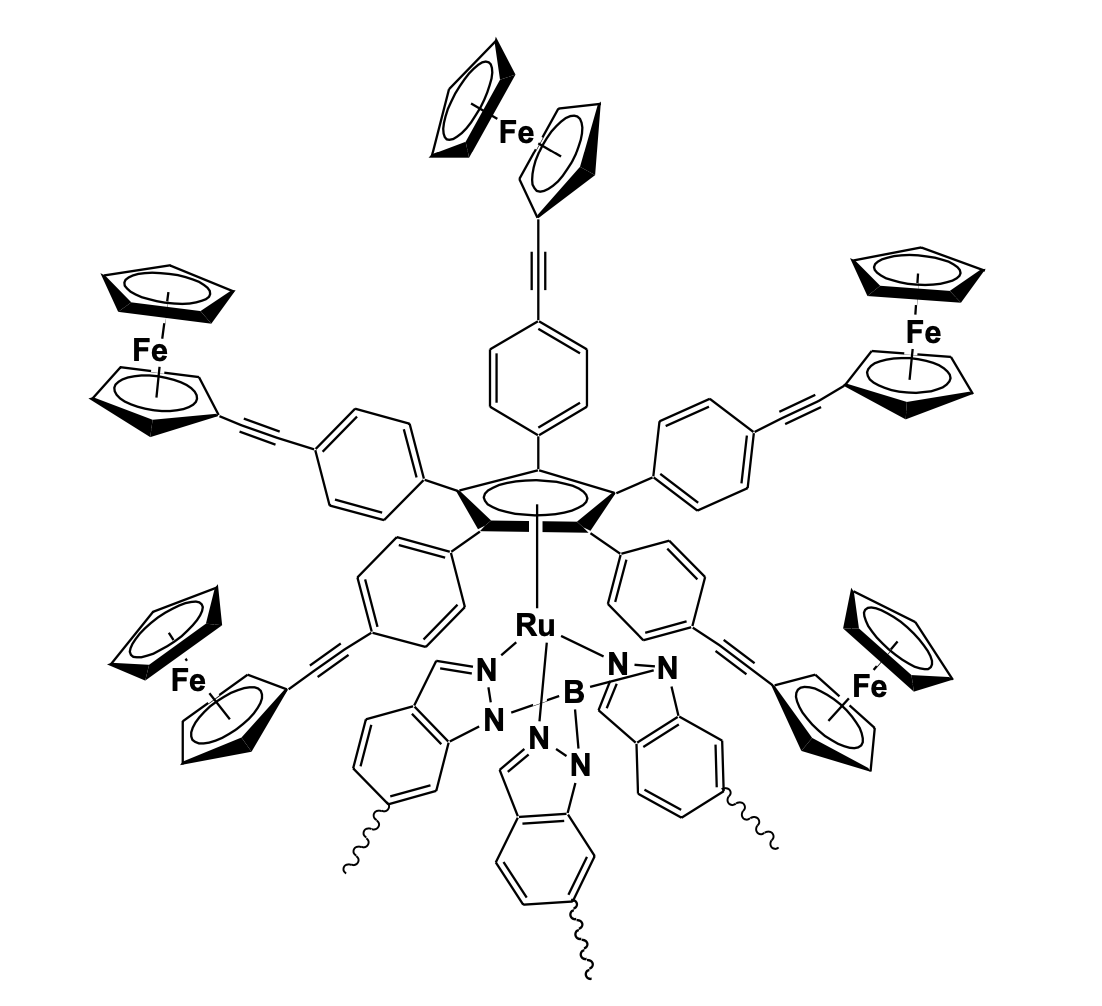
The synthetic procedure involves an olefination reaction by the Wittig method and a Pd-mediated C-C coupling reaction using a palladium pincer complex. The electrochemical analysis of the compound revealed two separate redox responses for the peripheral ferrocenyl units and the central ferrocenyl unit displaying distinct electronic coupling behavior between the two extreme redox species [49]. In a remarkable use of a multi-ferrocenyl system in miniaturized molecular devices, Rapenne and Launay reported the concept of an electrically fuel ed single molecular rotary motor based on the transport of electrons between two electrodes by the electroactive groups attached to a central rotatable core. The motor is based on a ruthenium complex with a piano stool geometry bearing two different ligands that act as a rotor and a stator. The stator is a tripodal hydrotris(indazolyl)borate ligand functionalized by ester groups while the rotor is a cyclopentadienyl (Cp) ligand connected to five ferrocenyl groups by linear and rigid arms (Figure 16) [50].
The cyclic voltammetry in dichloromethane with nBu4NPF6 supporting electrolyte displayed one reversible peak for ferrocenyl units while promising effects appeared on electron exchange which triggers a change in the molecular geometry. Penta- and hexa-ferrocenyl benzenes have been prepared by Negishi type ferrocenylation reaction of hexabromo- or hexaiodobenzene with diferrocenylzinc. The molecules are of great interest due to their electronically tunable dendritic substructure with potential in electronic, magnetic, optical, and catalytic applications and because of the presence of crowded arene type structures that may function as molecular gear. The single-crystal structural analysis of the hexa-ferrocenyl benzene revealed that the central benzene ring is not planer and adopts a chair conformation. The cyclic voltam-metric study showed three well-separated reversible 1e, 2e, and 3e redox couples sug-gesting effective electronic communication (Figure 17) [51].
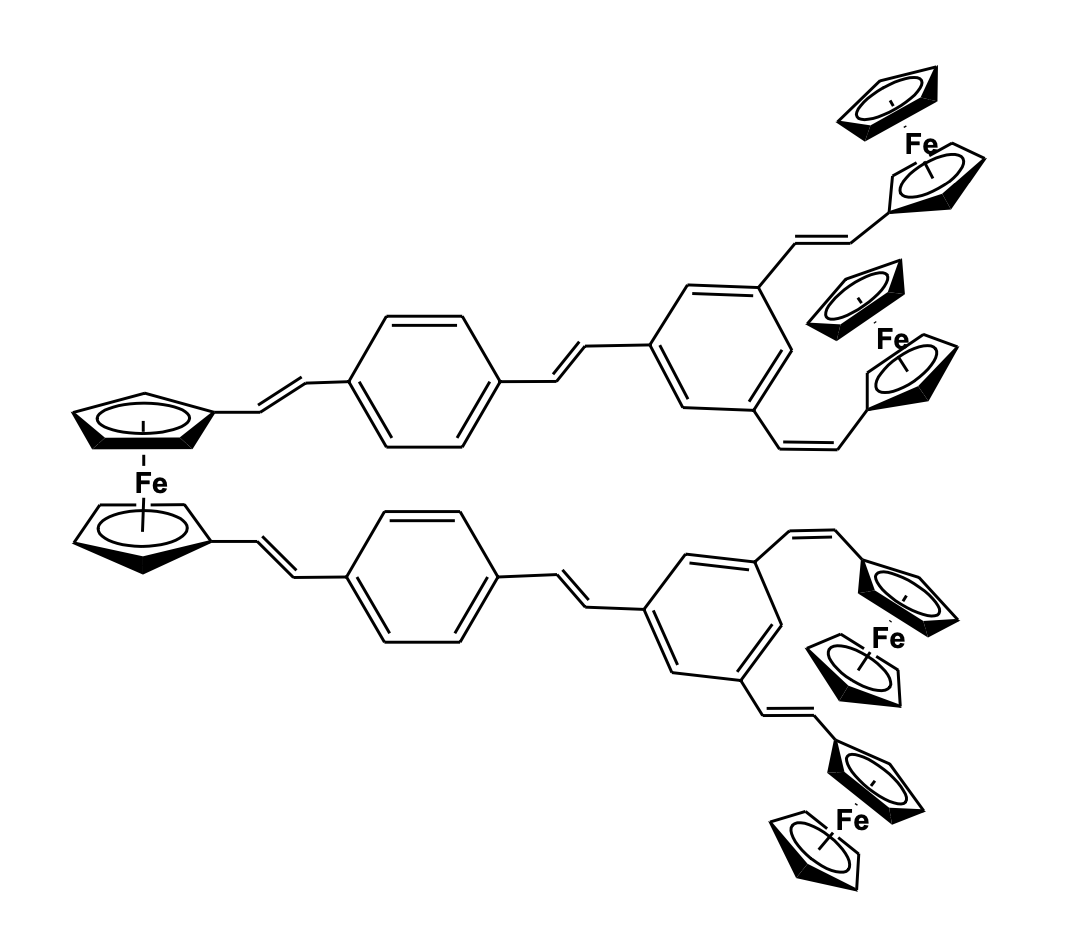
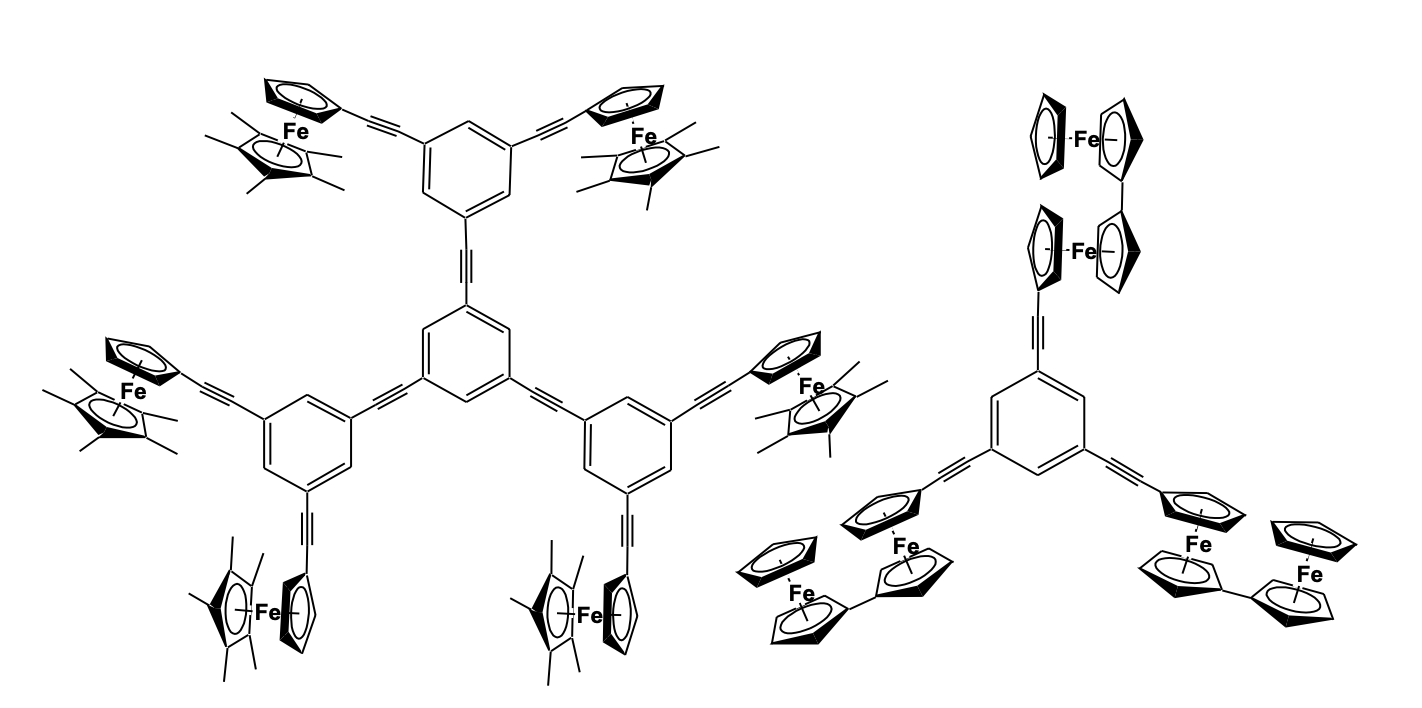
Electron transfer mechanisms in multi-redox systems like polymers, dendrimers, and macrocyclic moieties are of prime importance due to their relevance with biologically inspired electron transfer and redox processes. Also, these systems have been of interest for their potential as mixed-valence species, redox recognition, and catalysis. Ferrocenyl entities have always been a good choice to act as redox species due to their reversible process and stability of the mixed valence systems. Recently, the research group of Astruc has reported the synthesis of a series of rigid hexa- and dodeca- ferrocenyl arene centered poly(ethy-nylphenylene) dendrimers with C3 sym-metry using Sonogashira type coupling reaction and studied their electron-transfer properties. The polyferrocenyl entities showed distinct communication and stabilization of the mixed valence species (Figure 18) [52,53].
Conclusion Multinuclear and oligomeric ferrocene complexes have been extensively studied in the last decade for potential applications in multielectron redox catalysis, electron storage devices and as redox switchable molecules. Some of these multi-ferrocenyl systems can selectively vary their electronic properties by oxidation or reduction processes, making them important for minia-turized molecular switches and sensor devices. Therefore, this review summarizes the recent development of the multi-ferro-cenyl system and its synthetic strategies. Acknowledgments I very much want to thank Midnapore city college for providing me infrastructure to work and also I am thankful to my family for providing me mental support. References
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||