|
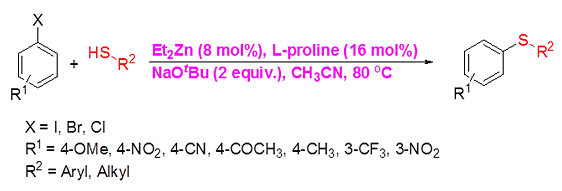
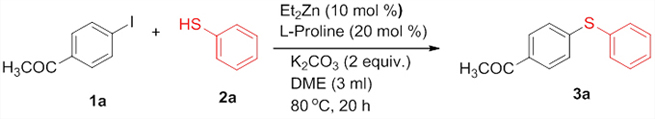
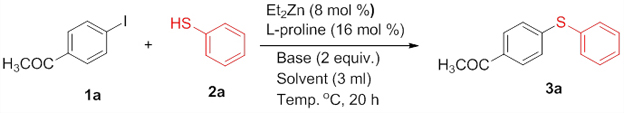
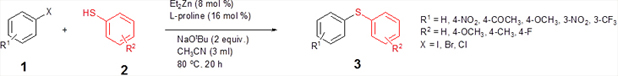
Introduction The construction of diarylthioether motifs has been a topic of immense interest for the past few years. This is due to the existence of the sulfide moiety in a large number of biologically and pharmacologically active compounds [1]. The presence of sulfur containing moieties in bioactive molecules offers subtle effects on their properties, as in the case of thio-nucleosides and thio-sugars [2]. There also exist a large number of commonly known antifungal agents with aryl sulfide moiety in their skeleton. Fenticonazole [3], ajoene [4], enediyne [5], and thiarubines [6] are some examples. A class of diaminodiphenylsulfones (DADS) [7] is well-known for their potent antibacterial activity against a variety of microorganisms. Diarylthioethers with heterocyclic motifs are the most commonly present structures in many drugs and are used for the treatment of diseases such as breast cancer [8], inflammatory diseases [9], HIV [10], and Alzheimer’s disease [11]. Owing to their great importance, a number of general methods are available for the synthesis of diarylthioethers. For example: the reduction of sulfones or sulfoxides, the coupling of metal thiolates and aryl iodides under elevated temperature, transition metal catalyzed cross-coupling reactions, etc. [12]. For the last few years, significant developments have been realized in the field of transition metal catalyzed carbon-heteroatom bond-forming reactions [13]. Out of this, numerous methodologies have been introduced for the carbon-nitrogen and carbon-oxygen bond-forming reactions while that for carbon-sulfur bond formation is moderate. This is due to the deactivation of the metal catalysts by organosulfur reagents. The organosulfur reagents deactivate the metal catalysts by forming strong coordinate bonds with active metal centers. However, a wide array of transition metal catalysts are successfully utilized for the carbon-sulfur bond-forming reactions, comprising of palladium [14], copper [15], nickel [16], cobalt [17], iron [18], rhodium [19], indium [20], and zinc [21]. The first report on transition metal catalyzed carbon-sulfur bond formation was published by Migita et al. in 1978 using catalytic amount of tetrakis (triphenylphophine) palladium complex [22]. Results & Discussion Recently, we have reported the first zinc-catalyzed C-S cross-coupling reaction of aryl halides with aryl and alkyl thiols using Et2Zn:L-proline catalytic system [21]. Herein, we report a detailed study on the zinc-catalyzed C-S cross-coupling reaction in presence of L-proline. We initiated the reaction by treating 4-iodoacetophenone (1a) with thiophenol (2a) in the presence of L-proline and K2CO3 in DME at 800C under nitrogen atmosphere (Scheme 1). After 20 hours of stirring, the solvent was removed under reduced pressure in a rotary evaporator and the product (3a) was separated from the crude reaction mixture by column chromatography on silicagel using EtOAc-hexane as the eluent to get a colourless solid.
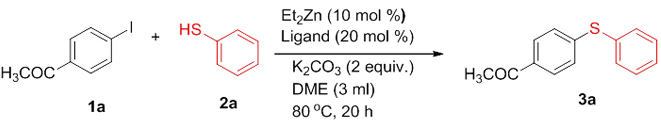
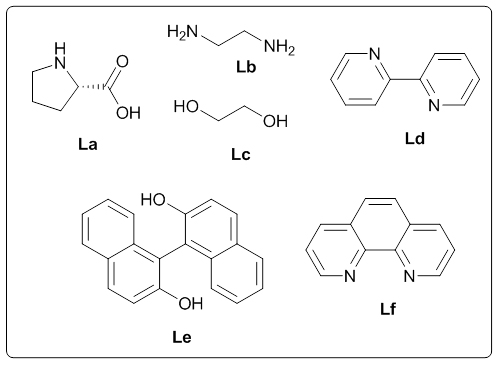
The structure of the product (3a) was established by nuclear magnetic resonance, mass spectrometric, and other analyses. The 1H NMR spectrum of the product showed a doublet (J = 8.4 Hz) for two protons at δ 7.83 corresponding to the two symmetric protons of the aromatic ring containing the acetyl group. The two protons appearing as a multiplet at δ 7.51-7.48 and the three protons appearing as a multiplet at δ 7.41-7.39 correspond to the protons of the unsubstituted aromatic ring. The doublet (J = 8.4 Hz) at δ 7.22 for two protons corresponds to the two symmetric protons of the aromatic ring containing the acetyl group. The three protons of the acetyl group appeared as a singlet at δ 2.55. In the 13C NMR spectrum, the carbonyl carbon resonated at δ 197.10 and the aromatic carbons resonated at δ 144.92-127.52. The methyl carbon resonated at δ 26.46. The IR spectrum showed an absorption at 3060 cm-1 corresponding to the aromatic C-H stretching while that at 1669 cm-1 is attributed to the C=O stretching. The C-S stretching appeared at 616 cm-1. The HRMS of the compound (3a) also matched well with the calculated value. Moreover, all the spectral data were in good agreement with the reported values [23]. After having characterized the product, we decided to conduct optimization studies for the reaction in detail. For this, the most commonly available and simple ligands, La-Lf, were chosen for screening.
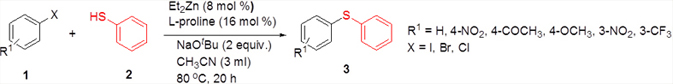
The ligand screening studies showed that L-proline (La) and 1,1’-bi-2-naphthol (Le) gave higher yield compared to the other ligands (Table 1: Entries 1, 5). Ligands such as 2,2’-bipyridyl (Ld) and 1,10-phenanthroline (Lf) gave lower amount of the product (Table 1: Entries 6, 4), while simple ligands like ethylenediamine (Lb) and ethyleneglycol (Lc) afforded only trace amount of the product (Table 1: Entries 2, 3). Based on the above observations, we decided to choose L-proline as the ligand for the zinc-catalyzed C-S cross-coupling reaction since it is simple, easily available, and eco-friendly. The catalyst loading studies were then conducted which showed that maximum yield was obtained when the catalyst loading was 8 mol% Et2Zn and 16 mol% of L-proline (Table 1: Entry 7). Further reduction of the amount of the catalyst reduced the yield of the product appreciably (Table 1: Entries 1, 7-10). After finding the optimal catalyst loading, we studied the influence of solvents in the coupling reaction. The results showed that both DME and acetonitrile are good solvents for this coupling reaction, since they gave comparatively good yield of the expected product (Table 2: Entries 3, 9-10, 14-16). In the presence of THF and tBuOH as the solvents, the coupling took place with low yield of the product (Table 2: Entries 4, 6). It was observed that the solvents such as DMSO, DMF, toluene, and 1,4-dioxane were not effective for the coupling (Table 2: Entries 2, 5, 7-8). Next, we examined the influence of bases using both DME and acetonitrile as the reaction solvent. After screening different bases, it was revealed that bases such as K2CO3, NaOtBu, Cs2CO3, and KOtBu were better for the reaction (Table 2: Entries 3, 9-10, 14-16). NaH gave a lesser amount of the desired product (Table 2: Entries 12-13), while the organic base Et3N did not afford any product (Table 2: Entry 11). Many attempts to make the reaction complete by increasing the temperature were unsuccessful; low yield of the product was observed at elevated temperature (Table 2: Entry 17). No product was obtained when the reaction was carried out at room temperature (Table 2: Entry 18). Employing 1.5 equiv. of NaOtBu as the base gave lower yield of the product (Table 2, Entry 19). When the reaction was performed without any base, no product could be observed (Table 2: Entry 20). Only traces of the product were detected in the absence of both Et2Zn and L-proline (Table 2: Entry 21). When the reaction was carried out in the absence of inert atmosphere, the required product was obtained only in trace quantity (Table 2: Entry 22). From all these observations, it was concluded that the overall optimal condition for the desired zinc-catalyzed C-S cross-coupling reaction was a combination of 1 equiv. of 4-iodoacetophenone, 1.1 equiv. of thiophenol, 2 equiv. of NaOtBu, 8 mol% of Et2Zn, and 16 mol% of L-proline in acetonitrile at 800C under nitrogen atmosphere (Table 2: Entry 16).
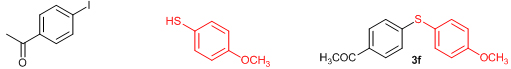
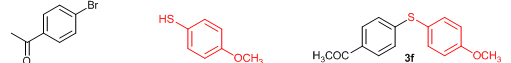
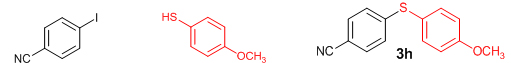
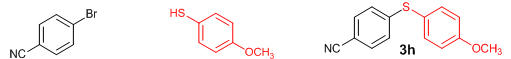
Next, we explored the generality of the zinc-catalyzed C-S cross-coupling reaction using the newly developed optimal conditions. The reaction between differently substituted aryl halides and thiophenol was tested and the results are summarized in Table 3. The presence of electron-withdrawing groups on the aryl iodides afforded the respective sulfides in excellent yields (3a, 3c, 3e). But the electron-donating methoxy group on the aryl halide significantly lowered the yield of the product (3d). To our delight, the reaction worked well in the case of unsubstituted aryl iodide with simple thiophenol (3b). The protocol was then applied to aryl bromides, which also gave the sulfide product; but in low to moderate yields (Table 3: Entries 2, 6, 8). The methodology was then extended to aryl chloride, viz., 4-chloroacetophenone which afforded 33% of the sulfide (Table 3: Entries 3). We also attempted the reaction of ortho-substituted aryl halides with aryl thiols, which did not give isolable quantity of the product presumably due to steric reasons; but the product formation was detected in GC-MS.
We then carried out the reaction between substituted thiophenols and aryl halides. It was observed that the electronic effects of the substituents on thiophenols have no considerable influence on the product yield. The thiophenol bearing electron-rich methoxy group at the C-4 position afforded the product with comparatively good yields (Table 4: Entries 1, 2, 4, 5). The thiophenol with methyl substituent at the C-4 position furnished moderate to good yield of the product (Table 4: Entries 7, 9, 11), while the C-4 fluoro-substituted thiophenol afforded good yields of the products (Table 4: Entries 12, 14). As anticipated, the substituted aryl bromides underwent cross-coupling reactions with 4-methoxy-, 4-methyl-, and 4-fluoro- substituted thiophenols affording the respective products in low yields (Table 4: Entry 3, 6, 8, 10, and 13). The meta-substituted aryl iodide gave only moderate yields of the product (Table 4: Entry 15).
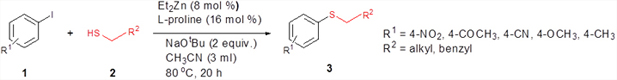
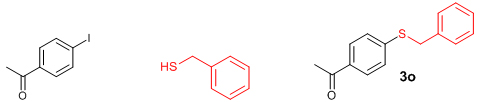
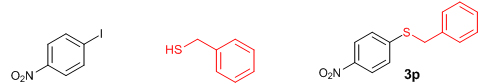
We then extended the reaction to alkyl thiols by conducting the reaction between aryl iodides and alkyl thiols under the optimal reaction conditions. As evident from Table 5, the protocol was tolerant towards many alkyl thiols. Benzyl thiol reacted smoothly with aryl iodides affording the desired product in good yield (Table 5: Entries 1, 2); but with 4-iodobenzonitrile as the coupling partner only moderate yield of the product could be obtained (Table 5: Entry 7). Notably, aliphatic thiols, such as n-butanethiol and i-propanethiol, also gave the corresponding products in moderate yields (Table 5: Entries 3-6). We also tried to extend this protocol to aryl iodides substituted with electron-releasing groups such as 4-iodotoluene and 4-iodoanisole; unfortunately, isolable quantity of the product could not be obtained although the product formation could be detected by GC-MS. In short, the new methodology works well in the case of aryl and alkyl thiols, including benzyl thiol with activated aryl iodides, allowing the facile preparation of a variety of sulfides in moderate to excellent yields.
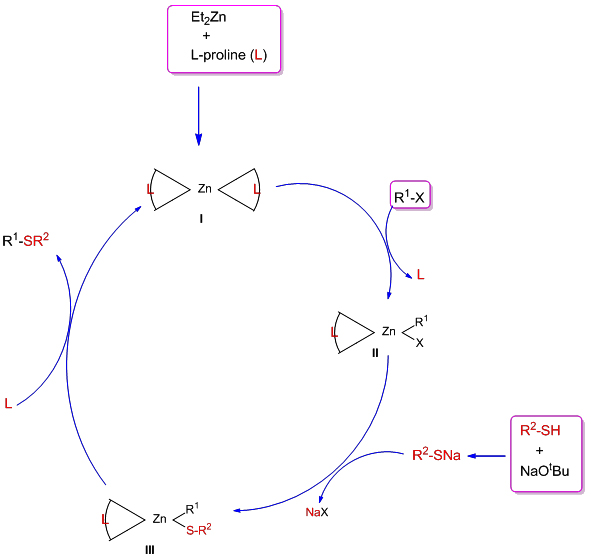
Even though a detailed study is necessary to unravel the mechanistic pathway of the novel zinc-catalyzed C-S cross-coupling reaction, we propose a tentative mechanism, as shown below (Scheme 2). The reaction between Et2Zn and L-proline would result in the in situ formation of a tetra-coordinated zinc-complex (I). This zinc-complex (I) can undergo oxidative addition with aryl halide by expelling one of the coordinated ligands, resulting in the generation of the Zn-complex (II). The complex (II) can then undergo ligand exchange with sodium thiolate, obtained by the deprotonation of thiophenol by NaOtBu, forming the complex (III). The reductive elimination of the zinc-complex (III) would afford the thioether with the regeneration of the complex-(I) and thus continues the catalytic cycle. The electron donating substituents on aryl halides significantly reduce the yield of the coupled product, presumably due to the sluggish oxidative addition of aryl halides with complex-(I). However, an alternative route via the coordination of thiolate anion with Zinc-proline complex followed by the oxidative addition of aryl halide cannot be completely ruled out.
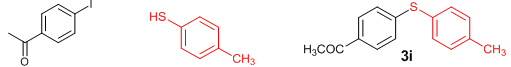
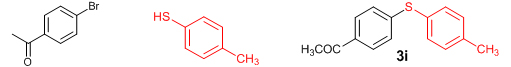
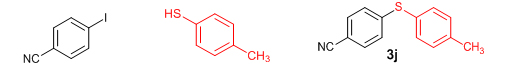
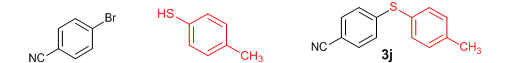
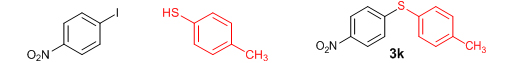
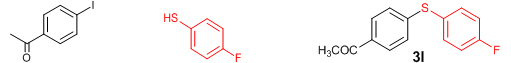
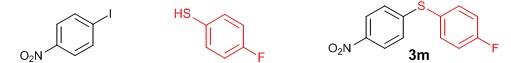
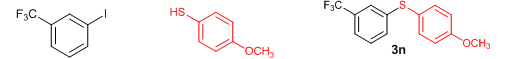
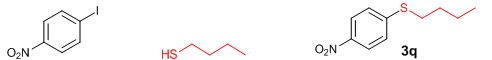
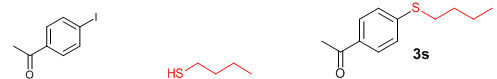
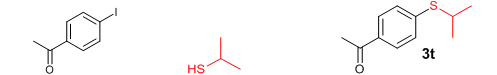
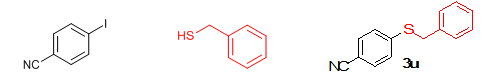
Conclusions In summary, we have developed an efficient and promising protocol for the zinc-catalyzed S-arylation of aryl and alkyl thiols with differently substituted aryl halides including iodides, bromides, and chlorides under mild reaction conditions. The in situ generated Et2Zn-proline system in CH3CN in the presence of NaOtBu at 800C showed very good catalytic activity in the C-S cross-coupling reactions. The versatility and environmental friendliness of this method, in addition to the high yields it provides, makes it viable for use in organic synthesis. The newly developed Zn-proline catalytic system is an efficient and successful combination for the production of aryl and alkyl sulfides in high yields with 8 mol % of catalyst loading, and shows high functional group tolerance. Experimental Section Typical experimental procedure for the synthesis of 1-(4-phenylsulfanylphenyl)ethanone (3a): A dry sealed tube was charged with 1 mmol (246 mg) of 4-iodoacetophenone, 16 mol% of L-proline (18 mg), and 2 equiv. of NaOtBu (192 mg) under nitrogen. To the above mixture was added 8 mol % of Et2Zn (1M in hexane, 0.08 ml) and 3 ml of acetonitrile followed by the addition of 1.1 mmol of thiophenol (0.11 ml) under nitrogen. The sealed tube was heated in an oil bath which was preheated to 80 oC and the reaction mixture was stirred under the same conditions for 20 hours. The reaction mixture was then cooled and extracted with ethyl acetate (3 x 15 ml) and the ethyl acetate layer was washed with saturated aqueous NaCl solution (1 x 15 ml). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure in a rotary evaporator. The crude residue was purified by column chromatography on silica gel using EtOAc-hexane as the eluent to get 217 mg (95%) of the product as a colourless solid. M. P: 670C; 1H NMR (400 MHz, CDCl3): δ 7.83 (d, J = 8.4 Hz, 2H), 7.51-7.48 (m, 2H), 7.41-7.39 (m, 3H), 7.22 (d, J = 8.4 Hz, 2H), 2.55 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 197.10, 144.92, 134.55, 133.87, 132.16, 129.69, 128.91, 128.79, 127.52, 26.46; IR (neat): 3060, 1669, 1555, 1182, 819, 616 cm-1; HRMS (QToF): [M+H]+ calculated for C14H13OS is 229.0687; found 229.0675 Diphenylsulfide (3b): Appearance: Colourless liquid; Yield: 149 mg (80%); HRMS (QToF): [M+H]+ calculated for C12H11S is 187.0781; found 187.0799; Spectroscopic data were identical to those published previously [24]. 4-Phenylsulfanylbenzonitrile (3c): Appearance: Yellow liquid; Yield: 205 mg (97%); HRMS (QToF): [M+H]+ calculated for C13H10NS is 212.0533; found 212.0526; Spectroscopic data were identical to those published previously [25]. 1-Methoxy-4-phenylsulfanylbenzene (3d): Appearance: Yellow liquid; Yield: 132 mg (61%); HRMS (QToF): [M]+ calculated for C13H12OS is 216.0609; found 216.0603; Spectroscopic data were identical to those published previously [24]. 1-Nitro-4-phenylsulfanylbenzene (3e): Appearance: Yellow solid; MP: 55-570C; Yield: 199 mg (86%); HRMS (QToF): [M+H]+ calculated for C12H10NO2S is 232.0432; found 232.0426; Spectroscopic data were identical to those published previously [26]. 1[4(4-Methoxyphenyl)sulfanylphenyl]ethanone (3f): Appearance: Colourless crystals; MP: 400C; Yield: 222 mg (86%); HRMS (QToF): [M+H]+ calculated for C15H15O2S is 259.0787; found 259.0783; Spectroscopic data were identical to those published previously [27]. 4-Nitrophenyl-4-Methoxysulfide (3g): Appearance: Pale yellow crystals; MP: 65-670C; Yield: 183 mg (70%); HRMS (QToF): [M+H]+ calculated for C13H12NO3S is 262.0532; found 262.0539; Spectroscopic data were identical to those published previously [18c]. 4(4-Methoxyphenyl)sulfanylbenzonitrile (3h): Appearance: Colourless solid; MP: 96-980C ; Yield: 207 mg (86%); HRMS (QToF) [M+H]+ calculated for C14H12NOS is 242.0639; found 242.0627; Spectroscopic data were identical to those published previously [27]. 1-(4-Tolylsulfanylphenyl)ethanone (3i): Appearance: Colourless solid; MP: 89-910C; Yield: 129 mg (53%); HRMS (QToF): [M+H]+ calculated for C15H15OS is 243.0840; found 243.0844; Spectroscopic data were identical to those published previously [25]. 4-Tolylsulfanylbenzonitrile (3j): Appearance: Colourless solid; MP: 100-1020C; Yield: 146 mg (65%); HRMS (QToF): [M+H]+ calculated for C14H12NS is 226.0684; found 226.0683; Spectroscopic data were identical to those published previously [28]. 4-Nitrophenyl-4-tolyl sulfide (3k): Appearance: Pale yellow solid; MP: 79-810C; Yield: 201mg (82%); HRMS (QToF): [M+H]+ calculated for C13H12NO2S is 246.0589; found 246.0588; Spectroscopic data were identical to those published previously [25]. 1-(4-Fluorophenylsulfanylphenyl)ethanone (3l): Appearance: Yellow liquid; Yield: 208 mg (85%); HRMS (QToF) [M+H]+ calculated for C14H12FOS is 247.0587; found 247.0589; Spectroscopic data were identical to those published previously [16d]. 1-Nitro-4-fluorophenylsulfanylbenzene (3m): Appearance: Pale yellow solid; MP: 97-990C; Yield: 210 mg (85%); HRMS (QToF): [M-H]+ calculated for C12H9NO2FS is 248.0181; found 248.0132; Spectroscopic data were identical to those published previously [16d]. 1-Methoxy-4-trifluoromethylphenylsulfanylbenzene (3n): Appearance: Clear liquid; Yield: 190 mg (67%); HRMS (QToF): [M]+ calculated for C14H11F3OS is 284.0482; found 284.0487; Spectroscopic data were identical to those published previously [15d]. 1(4-Benzylsulfanyl)phenylethanone (3o): Appearance: Colourless solid; MP: 110-1120C; Yield: 218 mg (90%); HRMS (QToF): [M+H]+ calculated for C15H15OS is 243.0844; found 243.0839; Spectroscopic data were identical to those published previously [15d]. 1-Benzylsulphanyl-4-Nitrobenzene (3p): Appearance: Yellow solid; MP: 97-990C; Yield: 196 mg (80%); HRMS (QToF) [M+H]+ calculated for C13H12NO2S is 246.0589; found 246.0588; Spectroscopic data were identical to those published previously [29]. 1-Butylsulfanyl-4-nitrobenzene (3q): Appearance: Pale yellow liquid; Yield: 125 mg (60%); HRMS (QToF) [M+H]+ calculated for C10H14NO2S is 210.0588; found 210.0545; Spectroscopic data were identical to those published previously [29]. 4-Butylsulfanylbenzonitrile (3r): Appearance: Pale yellow liquid; Yield: 107 mg (56%); HRMS (QToF) [M+H]+ calculated for C11H14NS is 192.0846; found 192.0836; Spectroscopic data were identical to those published previously [29]. 1(4-Butylsulfanyl)phenylethanone (3s): Appearance: Colourless liquid; Yield: 108 mg (52%); HRMS (QToF) [M+H]+ calculated for C12H17OS is 209.0994; found 209.0992; Spectroscopic data were identical to those published previously [15d]. 1-4-Propane-2-ylsulfanyl-phenylethan-1-one (3t): Appearance: Pale yellow liquid; Yield: 101 mg (52 %); HRMS (QToF) [M+H]+ calculated for C11H15OS is 195.0843; found 195.0849; Spectroscopic data were identical to those published previously [30]. 4-Benzylsulfanylbenzonitrile (3u): Appearance: Yellow liquid; Yield: 158 mg (70%); HRMS (QToF) [M+H]+ calculated for C14H12NS is 226.0684; found 226.0680; Spectroscopic data were identical to those published previously [16d]. Acknowledgements GA thanks the Kerala State Council for Science, Technology, and Environment (KSCSTE), Trivandrum, India (Order no. 341/2013/KSCSTE dated 15.03.2013) for financial support. APT, SKS, and KKK thank the KSCSTE-India, UGC-India and the Ministry of Social Justice and Empowerment, India for research fellowships, respectively. We thank the Inter University Instrumentation Centre (IUIC) and Institute for Intensive Research in Basic Sciences (IIRBS) of Mahatma Gandhi University for HRMS and NMR facilities, respectively. References
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||