|
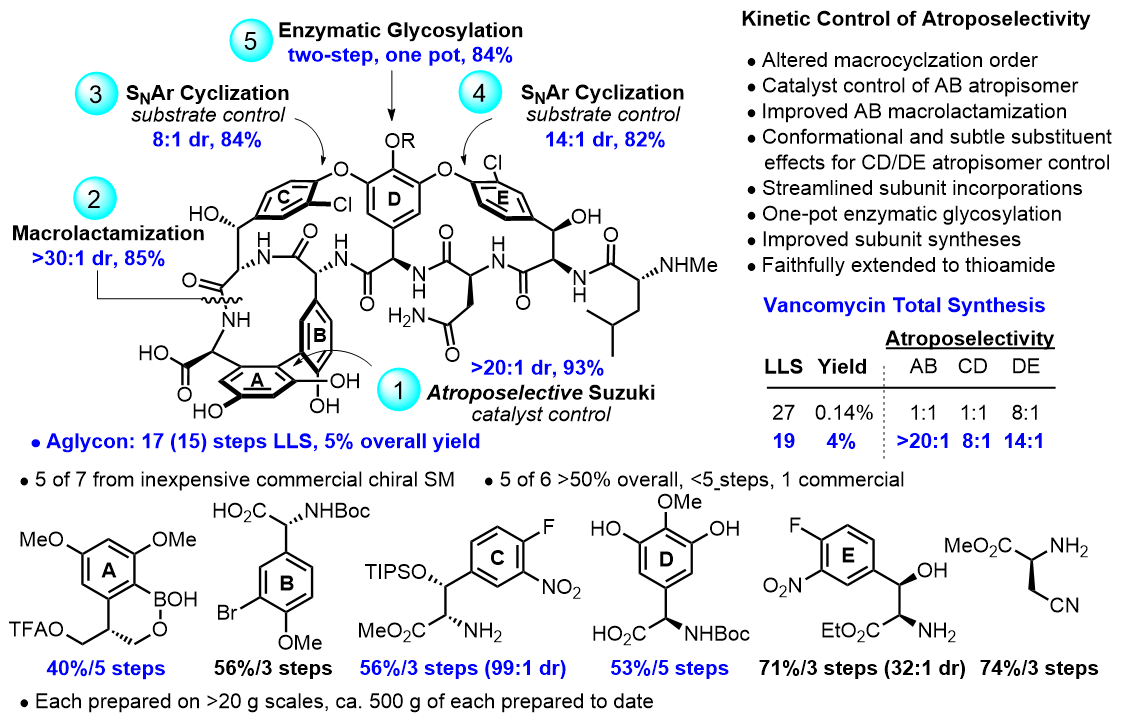
Introduction Natural products have provided and continue to define unique opportunities to address problems in biology and medicine. These opportunities have grown as the methods to identify their mechanisms of action and biological targets have improved and as the techniques used to directly determine their target bound structures have advanced [1]. With advances in organic chemistry and especially the total synthesis of complex natural products, the examination of key partial structures, compounds that contain deep-seated structural modifications, their unnatural diastereomers, and even their enantiomers provide a powerful complement to such studies [2–4]. Well- designed modifications in natural products may be used to address the structural basis of their interaction with biological targets, to define fundamental relationships between structure and activity, functional reactivity, and biological properties, or to mitigate liabilities. Because natural products integrate a constellation of properties into compact, highly-functionalized molecules, each structural feature and substituent are often thought to contribute to the biological activity. This is especially true when their targeted properties are directly related to their emergence in nature where they have undergone natural selection optimization. However, as highlighted herein, there are many examples where even a single atom change can substantially improve on their biological properties [5]. In such work, a challenging feature with natural products is to understand the subtle design elements integrated into their structures and then to rationally extend them to provide more selective, more potent, or more efficacious compounds. The studies are enabled by the development of divergent synthetic strategies and methods that advance the underlying systematic medicinal chemistry, targeting compounds bearing deep-seated structural changes not accessible by semisynthetic or biosynthetic means. In the work summarized, key structural modifications were designed to also improve defined properties, to endow the natural product with new properties or additional mechanisms of action, or to overcome intrinsic limitations of the natural product itself. The efforts provided supernatural products, a term introduced by my colleague, Ryan Shenvi, with properties superseding the parent natural product [6]. Although introduced with tongue and cheek humor by Shenvi just before Halloween, we have become enamored by the ability of the term “supernatural product” to describe such advances for not only experts in the field but especially for a general audience who may not appreciate the nature or magnitude of the accomplishments [7–9]. The design principles for creating the supernatural products highlighted include those that improve potency, increase selectivity, enhance durability toward raising resistance, broaden the spectrum of activity, improve chemical or metabolic stability, overcome limiting physical properties, add new additional mechanisms of action, enhance PK properties, overcome drug resistance, and/or improve in vivo efficacy. While some improvements may be regarded as iterative enhancements, others live up to their characterization as supernatural products. Naturally Occurring Cyclic Peptides Including the Glycopeptide Antibiotics A long-standing program has focused on the total synthesis and evaluation of naturally occurring biologically active cyclic peptides. The most recognized of the efforts are the total syntheses of naturally occurring glycolpeptide antibiotics including vancomycin and its aglycon, teicoplanin, ristocetin and ramoplanin aglycons, chloropeptin I and II, and the complestatins [10]. Along with these studies, we rationally redesigned the structure of vancomycin to achieve dual binding to both D-Ala-D-Ala and D-Ala-D-Lac, altering a single atom in the binding pocket such that it remains active against vancomycin-sensitive bacteria (e.g., MRSA), but is equally active against vancomycin-resistant bacteria (e.g., VRSA, VRE). The studies first defined the origin of the destabilized binding to the altered D-Ala-D-Lac target in resistant bacteria (100-fold derived from repulsive lone pair/lone pair interaction, 10-fold from lost H-bond) [11]. This led to the design and total syntheses of [Ψ[CH2NH]Tpg4]vancomycin and [Ψ[C(=NH)NH]Tpg4] vancomycin and their earlier aglycons, both of which displayed the dual ligand binding affinity and reinstated activity against vancomycin-resistant bacteria while maintaining activity against vancomycin sensitive bacteria. This was followed by the syntheses of peripherally-modified derivatives that we named maxamycins that: (1) contain a binding pocket and two synergistic peripheral modifications, (2) are endowed with three independent mechanisms of action only one of which is dependent on D-Ala-D-Ala binding, (3) display broad spectrum activity (e.g., MRSA, VanA/VanB VRSA and VRE) at remarkable potencies (MICs = 0.01 ㎍/mL), and (4) are durable antibiotics incapable of raising resistance in vitro and are potentially capable of extensive use for decades without fear of raising resistance [12,13]. With the discovery of the potent and durable activity of the maxamycins, α next generation total synthesis of vancomycin was developed that substantially improved synthetic access (Figure 1) [14].
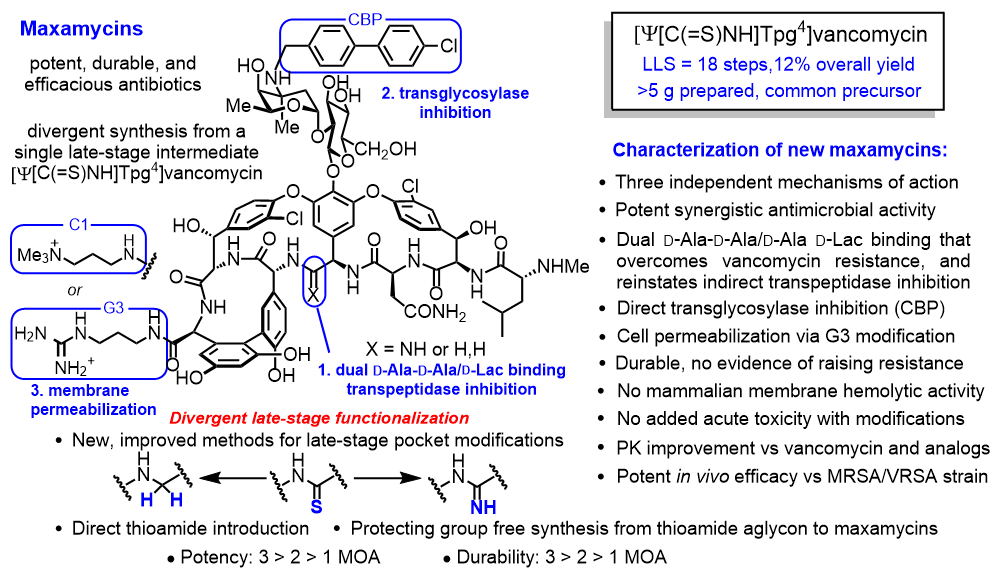
This divergent synthetic approach was subsequently further improved and implemented for the total syntheses of any maxamycin from a single late-stage intermediate, permitting traditional medicinal chemistry optimization on the complex glycolpeptide antibiotic [15]. The residue 4-thioamide of this late-stage intermediate, [Ψ[C(=S)NH]Tpg4]vancomycin, can be and was directly converted to any pocket-modified derivative that displays the dual ligand binding and paired with any combination of two different peripheral modifications, each introducing independent synergistic mechanisms of action without the use of protecting groups (Figure 2).
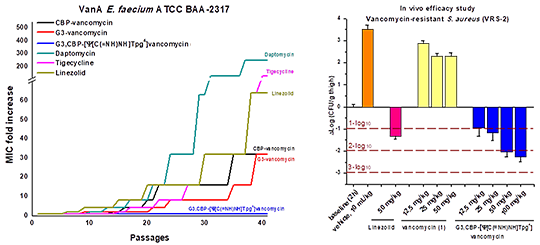
A class of true supernatural products that supersede the natural product, the maxamycins possess pocket modifications that overcome vancomycin resistance, reinstating transpeptidase inhibition and cell wall synthesis inhibition by binding to the altered target substrate D-Ala-D-Lac, while maintaining binding to the original target D-Ala- D-Ala of vancomycin. In addition, the peripheral modifications introduce two new synergistic mechanisms of action not seen with vancomycin itself. One peripheral modification (CBP modification) results in the direct competitive inhibition of transglycosylase affecting a separate step in cell wall biosynthesis and the second (G3 modification) induces cell permeability reducing cell wall integrity likely through cell wall teichoic acid binding and induced autolysin release, both of which are independent of one another and of D-Ala-D-Ala/D-Lac binding [15]. The synergic enhancement in activity derived from each modification, each of which increases both potency and durability against raising resistance, requires incorporation into a single molecule, indicating that it arises from the simultaneous expression of all three mechanisms of action at the same time and at the same place in the cell wall (Figure 3).
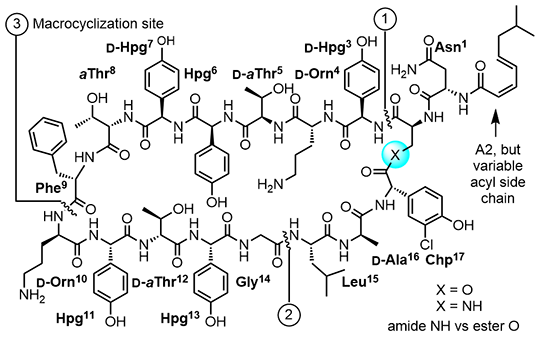
In the first such study, a prototypical maxamycin (G3,CBP-[Ψ[C(=N)NH]Tpg4]vancomycin) was shown to exhibit efficacious in vivo activity against the feared multidrug-resistant (MRSA) and vancomycin-resistant (VRSA) S. aureus bacterial strain (VanA VRS-2) for which vancomycin is inactive (Figure 3). Moreover, it was found that the C-terminus modifications (C1 and G3) offset the impact of hydrophobic vancosamine substitution, providing compounds with near ideal in vivo PK properties mitigating the short terminal half-life (0.5–1.3 h, t1/2), low exposure (AUC), inconsistent volume of distribution (Vd), and rapid clearance (CL) of vancomycin and the poor dose proportionality and problematic long terminal half-life of CBP-vancomycin [15]. A reduction in the complexity of the glycol-peptide structure through addition of two benign aryl chlorides that remove two synthetically challenging atropisomer elements (tetrachlorovancomycin [16] and the corresponding tetrachloromaxamycins [17]) permits an even more streamlined and scalable total synthesis and new opportunities for deep-seated core modifications. The ramoplanins are naturally occurring lipoglycodepsipeptides that are 2–10-fold more active than vancomycin against Gram-positive bacteria [18]. Ramoplanin A2 disrupts bacterial cell wall biosynthesis, inhibiting the intracellular conversion of lipid intermediate I to lipid intermediate II and the more accessible extracellular transglycosylase-catalyzed incorporation of lipid II into the glycan strand, steps that precede the site of action of vancomycin. Resistance to ramoplanin has not been detected, and cross resistance between ramoplanin and vancomycin has not been observed. Thus, it remains equally active against vancomycin-resistant organisms, in- cluding VanA/VanB VRE. Like vancomycin, ramoplanin acts by binding peptidoglycan precursors (lipid II > lipid I), sequestering these substrates from enzyme access, although the structural details of these interactions are not yet defined. In fact, ramoplanin embodies all the characteristics of vancomycin that contribute to its durability against resistance development. However, its instability derived from rapid depsipeptide hydrolysis precludes use for systemic infections and has limited its clinical exploration. Our development of the first and still only convergent total synthesis of the ramoplanin A1–A3 aglycons set the stage for its use in the preparation of key analogues. In these efforts, we demonstrated that synthetic [L-Dap2]ramoplanin A2 aglycon, which bears a linking amide in place of the sensitive depsipeptide ester in the backbone of the 49-membered macrocycle, is roughly 2-fold more potent ramoplanin A2 and its aglycon, and stable to 6 hydrolytic cleavage (Figure 4) [19].
Here, the single atom exchange in the compound does not impact the interaction of the natural product with its biological target or substantially alter its functional activity, but it mitigates its limiting metabolic instability. In our studies and on this stable amide template, an alanine scan of the complete structure was conducted (15 analogues prepared by total synthesis), establishing the impact and potential role of each residue and providing insights into the nature of its complex with lipid II [20]. Highlights derived from the alanine scan include: (1) the verification of the dominant role of Orn (10) (>500-fold reduction) consistent with an integral role in lipid II diphosphate bind- ing, (2) the more modest impact of Orn (4) (44-fold), suggesting that its role in binding lipid II is not as critical, (3) the disparate importance of each residue in a putative lipid II recognition domain proposed in early work (residues 3–10), (4) the significant impact (>20-fold) of nearly every residue in the dimerization domain (residues 11–14) later defined by Walker reflective of its greater importance, and (5) the lack of importance of the hydrophobic residues 16– 17 within the flexible loop that represents the membrane interacting domain (residues 15–17, 1–2). We also showed that the lipid side chain is essential for antimicrobial activity (200–800-fold reduction) and, in collaboration with Walker, showed it has no impact on lipid II binding or trans-glycosylase inhibition, indicating that its role is likely to anchor the antibiotic to the bacterial cell wall [19]. Complementing these studies on the stable amide-modified ramoplanin and other related studies, Walker used inhibition kinetics and binding assays to establish that ramoplanin preferentially inhibits the transglycosylase versus MurG catalyzed reactions of their substrates lipid II versus lipid I, that it exhibits a greater affinity for lipid II (KD = 3 nM) than lipid I (KD = 170 nM), and that it binds with a 2:1 stoichiometry consistent with functional dimerization [21]. Additional studies include efforts culminating in the total syntheses of the series of antitumor agents that act as DNA bisintercalators (sandramycin, luzopeptins A–C, quinoxapeptins A–C, thiocoraline, BE-22179, and triostin A), inhibitors of protein synthesis including cycloisodityrosine, deoxybouvardin, bouvardin/RA-VII, inhibitors of p53/MDM2 binding including chlorofusin and its diastereomers, as well as the natural products K-13, OF4949, piperazinomycin, HUN-7293, and streptide.
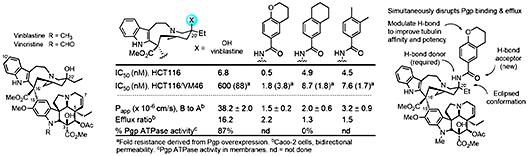
Vinblastine The discovery of vinblastine and its anti tumor activity led to the identification of tubulin as an especially effective oncology drug target [22]. Vinblastine binds at the tubulin α/β dimer−dimer interface where it destabilizes microtubulin assembly derived from the repetitive head-to-tail tubulin binding. Even by today’s standards, vinblastine and the related natural product vincristine are superb clinical drugs. They, and their biological target tubulin, remain the subject of intensive investigations because of their clinical importance in medicine, complex structures, low natural abundance, and unique mechanism of action. Following work that provided total syntheses of vinblastine and vincristine, our studies have probed the importance and role of the vindoline C16 methoxy group, C4 acetate, C5 ethyl substituent, C6–C7 double bond, and vindoline core itself, as well as the upper subunit C20’ ethyl substituent, C16’ methyl ester, and added C10’/C12’ indole substituents [23]. We defined the importance and anchoring role of the C20’ Et group and introduced the concept of “added benign complexity (ABC)” with an example that increased functional activity and tubulin binding affinity 10-fold. These and related studies showed that essentially every feature of the vinblastine core structure, every functional group and every substituent productively contributes to its properties. Unlike the removal of structural features or substituents that have a detrimental impact, the additions of new structural features not found in the natural product have been found that enhance target tubulin binding affinity and functional activity while simultaneously disrupting Pgp binding, transport, and functional resistance. Enabled by methodology that we introduced, a series of previously inaccessible vinblastine C20’ modifications were disclosed that display remarkable properties: 20’ ureas (30-fold increase in potency, reduced resistance due to Pgp efflux) [24], ultra-potent 20’ ureas (stunning 100-fold increase in potency derived from further disruption of the tubulin protein–protein interaction) [25], and vinblastine 20’ amides that completely overcome and mitigate resistance derived from Pgp overexpression and drug efflux (Figure 5) [26].
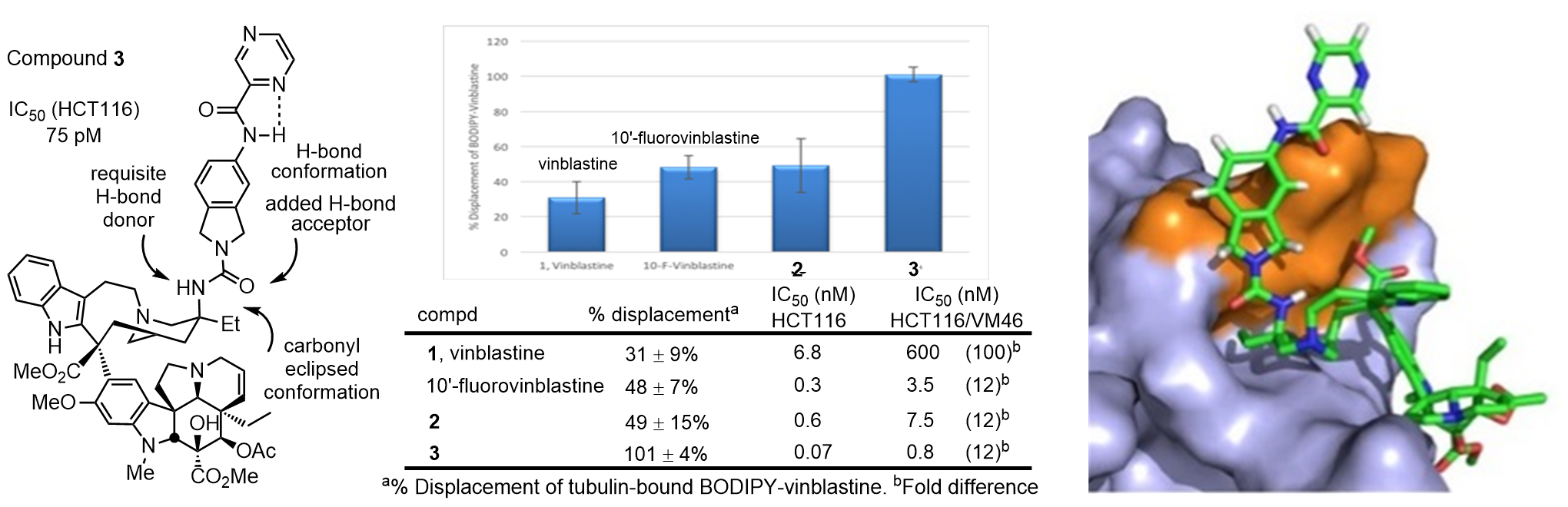
These studies were based on well-founded rationales, used systematic well-designed analogues (>400 analogues) to answer fundamental questions on the molecular interaction of vinblastine with its biological target (tubulin) or targeting its source of resistance (Pgp), enlisted traditional medicinal chemistry correlations to optimize activities, were accessed by new methodology developed for this purpose, and relied on a series of key functional and on-target assays to produce supernatural products far exceeding the potency and properties on the natural product. Approaches to improve the biological properties of natural products often strive to identify the essential pharmacophore to simplify the structure or make functional group changes to improve biological target affinity or functional activity, change physical properties, enhance stability, or introduce conformational constraints. Aside from accessible semi-synthetic modifications of existing functional groups, rarely does one consider using chemical synthesis to add molecular complexity to the natural product. In part, this may be attributed to the added challenge intrinsic in the synthesis of an even more complex compound. We discovered synthetically-derived, structureally more complex vinblastines inaccessible from the natural product itself that are a stunning 100-fold more active (IC50’s 50–75 pM vs 7 nM, HCT116) [25], and that are now accessible because of advances in the total synthesis of the natural product (Figure 6).
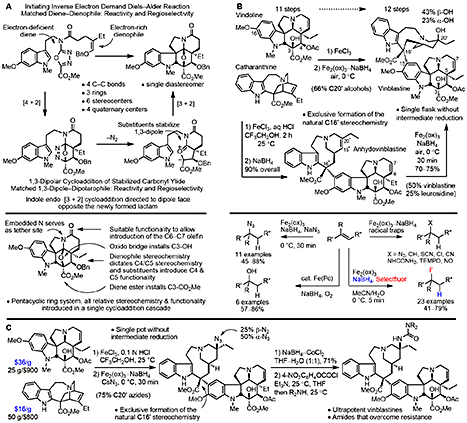
The newly discovered ultra-potent vinblastines bind tubulin with much higher affinity and further disrupt the tubulin head-to-tail α/β dimer–dimer interaction by virtue of the strategic placement of an added conformationally well-defined, rigid, and extended C20’ urea along the continuing head-to-tail tubulin α/β dimer−dimer interface. In this case, the added molecular complexity was used to markedly enhance target binding and functional biological activity (100-fold) [25]. In these efforts, vinblastine served as the inspiration for the discovery of a powerful intramolecular 1,3,4-oxadiazole cascade [4+2]/[3+2] cycloaddition reaction for the synthesis of the vindoline-derived lower subunit [27], development of a diastere-ospecific Fe(III)-mediated biomimetic coupling of vindoline with catharanthine to pro-vide anhydrovinblastine [28–30], and dis- covery of a powerful free radical hydrogen atom transfer functionalization of alkenes (Fe(III)–NaBH4/radical traps, [31,32]) that permits the in situ C20’ functionalization used to install the vinblastine C20’ alcohol or corresponding amine for amide and urea derivatization, stimulating further developments in the MHAT field (Figure 7).
Over 400 analogues of vinblastine were prepared, and it is a tribute to the advances in organic synthesis that such work can now be conducted on a natural product of a complexity once thought refractory to such an approach. Although it could not have been imagined at the stage that we initiated our efforts, the C20’ vinblastine analogues are now available in three steps from inexpensive commercially available materials (catharanthine, $16/g; vindoline, $36/g; Figure 7) [25].
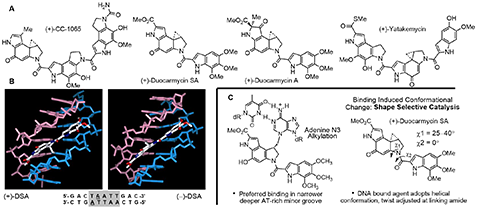
DNA Binding Natural Products: Total Synthesis, DNA Recognition, and Biological Properties An early focus of our work targeted DNA binding, alkylation, and cleaving agents that exhibit antitumor activity [33,34]. These studies include work on a class of natural products composed of CC-1065, duocarmycin A and SA, and yatakemycin where we not only conducted total syntheses of each natural product [35–37], defining the absolute stereochemistry and correcting a misassigned structure (yatakemycin), but also characterized their DNA alkylation properties [38–42] (Figure 8).
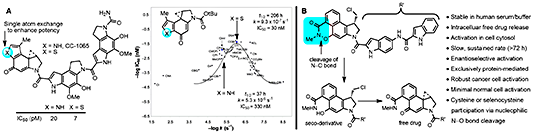
In these studies, we defined their DNA alkylation selectivity including that of their biologically active unnatural enantiomers, established their alkylation rates, reversibility, stereoelectronically-controlled reaction regioselectivity, and characterized their adenine N3 adducts. We defined the source of their DNA alkylation selectivity (non- covalent AT-rich binding selectivity – shape selective recognition [43]) and identified the source of catalysis for the DNA alkylation reaction (DNA binding induced conform- ational change disrupting the stabilizing vinylogous amide conjugation – shape dependent catalysis [44,45]). We provided high-resolution NMR-derived structures of the natural products bound to DNA [46–48] and established that they are subject to an exquisitely selective “target-based activetion”. More than 2000 analogues of the natural products that contain deep-seated structural changes have been disclosed in our work and were used to define the relationships be- tween structure and reactivity or structure and activity [49], and their contributions to the expression of the DNA alkylation properties and biological activity of the natural products (e.g., “hydrophobic binding-driven-bonding” [50] and a predictive parabolic relationship between reactivity and biological potency [51]). A compilation of the data derived from more than 30 deep-seated modifications in the alkylation subunit, many of which entailed single atom changes, resulted in the establishment of the predictive parabolic relationship between the alkylation subunit reactivity and the resulting cell growth inhibition potency that spanned a 104−106 range of reactivity and activity[51](Figure9).
Presumably, this fundamental relationship reflects the fact that the compound must be sufficiently stable to reach its biological target yet remain sufficiently reactive to alkylate DNA once it does. The parabolic relationship defined this optimal balance between reactivity and stability, providing a fundamental design feature that was used to improve the potency of CC-1065 by a single heavy atom exchange [52] (Figure 9). Our recent work led to the discovery of a new class of reductively activated prodrugs specifically designed for this class of natural products, mapping seamlessly onto their core structures [53]. This new class of prodrugs proved especially efficacious in vivo, yet are remarkably non-toxic, displaying no myelotoxicity and effectively taming the potent cytotoxic activity of the natural products (Figure 9). Notably, the prototype members of the prodrug class bear structural simplifications that emerged in our studies that improve stability, enhance in vitro potency and in vivo efficacy, and simplify synthetic access. Ongoing preclinical studies of one of our lead compounds defined the site (intracellular, cancer vs normal cell lines), slow sustained rate, and mechanism of the exclusively protein-mediated prodrug cleavage and free drug release mediated by protein cysteine residue[54]. Optimization of these compounds fortolerability, safety, and therapeutic efficacymay provide a breakthrough in the clinicaltreatment of cancer, defining a new targeted precision therapy. Bleomycin is a clinically employed antitumor drug that derives its properties through the sequence-selective cleavage of DNA in a process that is both metal ion andO2 dependent [55−57]. Through development of a modular synthesis capable ofmodifying each region of the molecule, seminal studies (ca. 70 analogues) that probed each substituent and each subunit in the structure confirmed the origin of DNA cleavage selectivity derived from G triplex-like H-bonding in the minor groove, defined fundamental conformational properties ofthe linker region contributing to the efficiency of DNA cleavage, identified a conformational swivel point accounting for double-strand DNA cleavage from a singlebound site, and provided a high-resolution NMR-derived structure of DNA bound deglycobleomycin A2 [58,59] (Figure 10).
Combined, the studies helped define a role not only for each subunit, but also the importance of each substituent in this remarkable molecule. In the course of our work, we also prepared a >9000-membered screening library of distamycin analogues [60], discovered and defined the DNA cross-linking properties of ischrysohermidin and established the origin of its selectivity [61], and studied the naturally occurring bis-intercalators (sandramycin, luzopeptins, BE-22179, quinoxapeptins, thiocoraline), defining their DNA binding selectivity, its origins, kinetics of binding, and established a high-resolution NMR-derived structure of sandramycin bound to DNA [62]. In these studies, we introduced a powerful fluorescent intercalator displacement (FID) assay for establishing DNA binding selectivity or affinity [63,64] that complements DNA footprinting and developed a convenient M13-derived alternative to 32P-end-labeling of restriction fragments for DNA cleavage studies[65].
Natural Products Total Synthesis, New Synthetic Methodology In our work, more than 100 natural products have been prepared by total syntheses, of which most represent biologically active natural products chosen by virtue of their properties. Their structures inspired the development of new synthetic methodology designed for the natural products of interest. Many were first total syntheses, defining or correcting the stereochemistry or structure, and often constitute concise, efficient total syntheses easily identifiable with our efforts. Highlights, including those that subsequently probed or defined structure–function properties not described elsewhere, targeted the total syntheses of streptonigrin (1983), juncusol (1984), rufescine and imelutine (1984), colchicine (1985), lavendamycin (1985), PDE-I and PDE-II (1987), prodigiosin and prodigiosene (1988), trikentrin A (1991), combretastatin (1991), (–)-pyrimidoblamic acid (1993), streptonigrone (1993), isochrysohermidin (1993), (+)-P-3A (1994), fredericamycin (1995), grandirubrine and imerubrine (1985), nothapodytine B and (–)-mappicine (1998), ningalin A, lamellarin O, lukianol A, and storniamide A (1999), phomazarin (1999), ningalin B (2000), distamycin (2000), hippadine (2000), rubrulone (2000), fostriecin (2001), (–)-roseophilin (2001), (+)-camptothecin (2002), anhydrolycorinone (2002), minovine (2005), piericidin A1 and B1 (2005), ningalin D (2005), (–)-vindorosine (2006), (–)-vindoline (2006), N-Methylaspidospermidine (2006), cytostatin (2006), phostriecin (2010), (+)-fendleridine and (+)-acetylaspidoalbidine (2010), (–)-vindorosine (2010), lycogarubin C and lycogalic acid (2010), (–)-aspidospermine (2012), (+)-spegazzinine (2012), kopsinine (2013), asymmetric synthesis of (+)-P-3A and (–)-pyrimidoblamic acid (2014), (–)-kopsifoline D (2014), (–)-deoxoapodine (2014), (–)-kopsinine (2015), dihydrolysergol and dihydrolysergic acid (2015), methoxatin (2016), streptide (2019), meayamycin (2020), (–)-pseudocopsinine (2020), (–)-minovincinine (2020), (–)-strempeliopine (2021), and (+)-paucidactine D (2025). In this work, new synthetic methodology or synthetic strategies have been introduced. Most recognized of these efforts include the inverse electron demand Diels–Alder reactions of heterocyclic azadienes [66], including the first reported organocatalytic Diels–Alder reaction, the first general method for catalyzing such reactions (solvent H-bonding of HFIP), the successful use of 1,2,3-triazine cycloaddition reactions utilizing powerful substituent effects, and the first synthesis and cycloaddition reactions of 1,2,3,5-tetrazines [67]. We introduced and developed the cycloaddition reactions of acyclic 1-azadienes (N-sulfonyl-1-azabutadienes), identified a unique transition state anomeric effect responsible for their remarkable endo diastereoselectivity, and developed a powerful asymmetric variant of the reactions [66]. We introduced and developed the powerful cascade [4+2]/[3+2] cycloaddition reactions of 1,3,4-oxadiazoles [27], and pioneered the cycloaddition reactions of cyclopropenone ketals ([4+2] cycloaddition) including the discovery of their reversible thermal (80°C) ring opening to p-delocalized singlet vinylcarbenes and subsequent substrate dependent cycloaddition reactions [66] ([1+2], [3+2], and [4+3] cycloadditions). We introduced the use of acyl radical generation from phenyl selenoesters and subsequent intermolecular and intramolecular alkene addition reactions [68], pioneered key palladium-catalyzed reactions including the first examples of a Pd(0)-mediated free amine amination of an aryl halide [69] in 1984, the conduct of unsymmetrical 2,2’-bipyrrole coupling via electrophilic Pd(II) C–H activation [70] in 1987, and introduced an intramolecular Pd(0)-catalyzed indole annulation for (macro)cyclizations [71]. We discovered and disclosed powerful free radical hydrogen atom transfer (HAT) functionalization, cyclization, or reduction reactions of unactivated alkenes (Fe(III)–NaBH4 with radical traps [31]) that has since gained widespread study. We defined the concept of divergent total synthesis [72] in 1984.
Conclusions Enabled by the development of divergent synthetic strategies and methods that advance the underlying systematic medicinal chemistry, the discovery of natural product analogues bearing deep-seated structural changes not accessible by other means is highlighted. Many display biological properties that not only improve those found in the natural product or overcome an intrinsic limitation, but have also been designed to endow them with productive new properties and even additional new mechanisms of action not expressed by the natural product, providing supernatural products.
Acknowledgments Many thanks to the talented group of pastand current coworkers for the conduct of the studies and the innovative ideas they introduced. The financial support of the National Institutes of Health (CA041101,CA042056, DA015648) is gratefully acknowledged.
References
|
||||||||||||||||||||||||||||||||||||||||||||||||