|
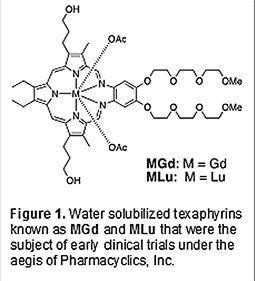
Introduction Texaphyrins are a class of pentaaza expanded porphyrins that were invented by members of the Sessler research group in the late 1980s [1-4]. They were seminal in their time in that they were the first analogues of porphyrins with an expanded central core that were documented as forming stable, non-labile 1:1 metal complexes with larger cations (i.e., those with ionic radii greater than accommodated within the core of porphyrins and related tetraaza macrocycles). Texaphyrins also garnered interest early on because they were aromatic systems, possessing a 22 π-electron periphery. As such, they led to a reassessment of the canonical thinking that aromaticity effects began to wane within systems containing more than 18 π-electrons. Both the coordination chemistry of texaphyrins and their electronic features were viewed as attractive in terms of potential technological development. In particular, they were found to form stable complexes with Lu(III) and Gd(III), both of which were characterized by lowest energy bands >700 nm in their respective UV-visible absorption spectra. The Lu(III) complex proved diamagnetic and a good singlet oxygen photosensitizer. In contrast, the Gd(III) complex was found to enhance so-called T1 relaxation effects. Perhaps not surprisingly, therefore, these two complexes attracted early interest as photodynamic therapy (PDT) [5,6] and magnetic resonance imaging (MRI) agents, respectively [7,8]. The further finding that the texaphyrins were 1) relatively easy to reduce and then 2) readily re-oxidized by oxygen to 3) produce reactive oxygen species made them of interest as potential radiation sensitizers [9-13]. Efforts to test the above promise led to the founding of Pharmacyclics, Inc. by Richard A. Miller, MD and the author in 1991 and the creation of water solubilized texaphyrin lutetium and gadolinium complexes referred to as motexafin lutetium (MLu) and motexafin gadolinium (MGd), respectively (Figure 1). Both complexes entered clinical trials under the aegis of Pharmacyclics, Inc. with the greatest development effort focused on MGd [14].
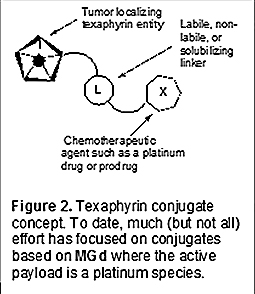
Unfortunately, in spite of progressing through a Phase III trial in the mid-2000s as a possible radiation sensitizer for metastatic lung cancer, MGd did not receive FDA approval due to the trial not reaching statistical significance in its endpoint. On the other hand, the agent was found to be well tolerated by patients at the administrated dosage levels [15,16]. In addition, based on both MRI studies and limited patient biopsies, it was found that MGd localized well to solid tumors [17-19]. This led to the consideration that MGd could be strategically utilized as a carrier for active anticancer agents that lack appreciable cancer targeting ability. The basic strategy that then began to evolve, shown in Figure 2, is that MGd would be functionalized in such a way that it could be linked to a specific chemotherapeutic so as to produce a conjugate that embodied the combined benefits of 1) targeting (due to the texaphyrin core) and 2) potency (due to the anticancer effect of the chemotherapeutic).
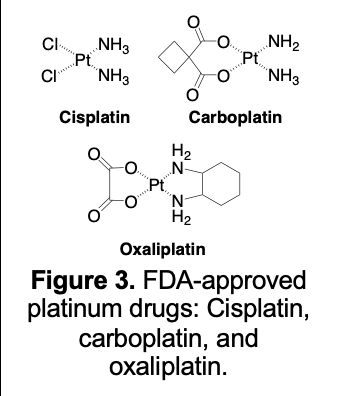
While early on a number of possible therapeutic payloads were considered [20-24], most of the effort within our texaphyrin conjugate program has focused on using functionalized MGd as a carrier for an active platinum agent. It is the evolution of the latter program that is the subject of this retrospective report. Currently, three Pt(II) complexes have been approved as cancer chemotherapeutics by the US FDA, namely cisplatin, carboplatin, and oxaliplatin (Figure 3). Together, these three agents remain part of the treatment regimen for roughly 50% of patients receiving cancer chemotherapy worldwide. Sadly, however, in many cases the administered platinum drugs are not curative. The FDA platinum drugs are noted for their lack of cancer targeting (thought responsible in part for systemic toxicity effects), as well as poor uptake/retention into cancer cells. These problems are exacerbated in the case of platinum resistance.
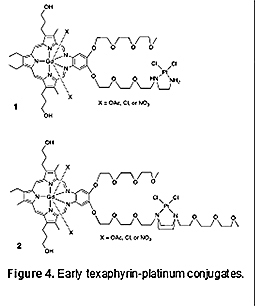
The determinants of platinum resistance are multifactorial. However, as the result of an incipient collaboration with Dr. Zahid Siddik of the MD Anderson Cancer Center we came to appreciate that resistance mechanisms include both pharmacological mechanisms (e.g., decreased drug uptake, increased GSH, and increased DNA adduct repair) and molecular mechanisms of resistance (e.g., a loss of the tumor suppressor protein 53 (p53) function, an increase in survivin, and an increase in B-cell lymphoma 2 (Bcl-2)) [25,26]. It was thought that developing texaphyrin-platinum conjugates could mitigate some of these clinical limitations. In collaboration first with Dr. Darren Magda and other researchers then at Pharmacyclics, Inc., and subsequently with Dr. Siddik, a program was thus launched to create and test platinum-texaphyrin conjugates. This effort can be divided into three phases, namely early exploratory efforts, preparation of first generation systems, and development of second generation systems. As detailed below, this body of work has culminated in the selection of OxaliTEX-Pt(IV) as OncoTEX's initial drug lead. Early Efforts to Create Texaphyrin-platinum Drug Conjugates The first texaphyrin-platinum conjugates were reported in U.S. Patent no. 6,207,660 that issued in March of 2001 [27]. The first summary of our texaphyrin-conjugate program in the chemical literature was published in 2004 [20]. Included in the latter report was the synthesis of two texaphyrin-platinum(II) conjugates, 1 and 2, based on the MGd core (Figure 4).
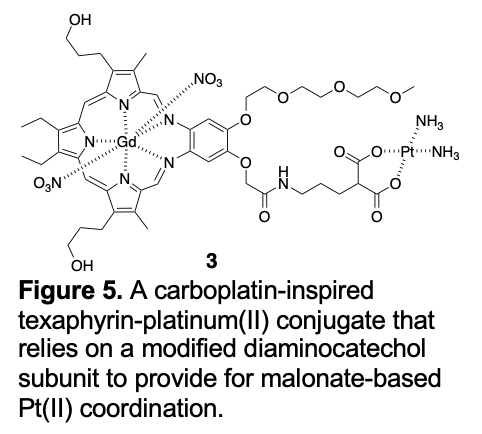
Unfortunately, neither conjugate proved appreciably soluble, either in aqueous media or organic solvents. This precluded a detailed study of these systems. An effort was thus made to redesign the synthetic approach. First Generation Texaphyrin-platinum Drug Conjugates Inspired by carboplatin, a malonate-based Pt(II) conjugate 3 was prepared (Figure 5).
This synthetic effort, reported in 2009 by Arambula, et al., provided the first generation of texaphyrin-platinum system suitable for biological testing [28]. As true for 1 and 2, compound 3 was prepared by modifying the diaminocatechol portion of the gadolinium(III) texaphyrin core. The combined presence of both carboxylate chelators and free hydroxypropyl "arms" of the texaphyrin core was found to impart aqueous solubility sufficient to allow for initial testing. First, hydrolysis-based Pt(II) release, presumably as the active diamino diaquo complex, Pt(NH2) 2(H2O)2, from the malonate chelator, was studied. A half-life for this release in phosphate-buffered saline was found to be ca. 72 h, a value that is commensurate with the rate of carboplatin hydrolysis [29]. Conjugate 3 was then tested in vitro using a standard tetrazolium dye (tetrazolium dye = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; MTT) reduction assay on several cell lines. In a wild type ovarian cancer cell line, A2780, antiproliferative potency analogous to carboplatin was observed (IC50 = 1.4 mM for 3 vs 1.6 mM for carboplatin). On the other hand, in an isogenic platinum resistant cell line, 2780CP, conjugate 3 proved ca. 2x more potent than carboplatin (IC50 = 14.4 mM for 3 vs 26.3 mM for carboplatin). However, MGd itself proved relatively effective (IC50 = 13.7 mM) in treating the 2780CP cell line, as did the combination of MGd and carboplatin (IC50 = 11.6 mM). It was noted that conjugate 3 provided for the formation of a greater number of DNA-platinum adducts as compared to various controls [30]. As such, conjugate 3 was deemed effective at overcoming mechanisms of resistance involving the effective delivery or retention of platinum to cancer cells. However, this conjugate was not found capable of overcoming the DNA damage tolerance that is endemic to carboplatin treatments of platinum resistant cancers. An effort was thus made to create a texaphyrin-platinum conjugate capable of overcoming these latter resistance mechanisms.
Oxaliplatin, in contrast to cisplatin and carboplatin, is known to reactivate dormant p53 [31]. It was thus considered likely that replacing the nitrogenous ligands about the Pt(II) center in conjugate 3 with the diaminocyclohexyl (DACH) subunit present in oxaliplatin might serve to increase the ability to overcome platinum resistance. This led to the preparation of conjugate 4 (Figure 6) as reported by Arambula, et al. in 2012 [32]. As true for 3, conjugate 4 proved capable of effectively delivering platinum to both wild type (A2780) and platinum resistance (2780CP) ovarian cancer cells, as determined by flameless atomic absorption spectroscopy (FAAS) analyses of the intracellular platinum concentrations. For conjugate 4, no difference between the cell lines was seen within error. Relative to oxaliplatin, conjugate 4 demonstrated a ≥2- and ≥4-fold increase in platinum uptake for the A2780 and 2780CP cell lines, respectively. However, despite the high potency of 4 for both cell lines, only a modest level of DNA-Pt adduct formation was observed. For example, it was found that the intracellular Pt content in A2780 cells was equal for treatments involving 4 and cisplatin; however, the DNA-Pt adducts from cisplatin was found to be ~4-fold higher relative to 4. This led us to infer that 4 might be participating in anticancer mechanisms similar to that of oxaliplatin that are independent of DNA damage [34].
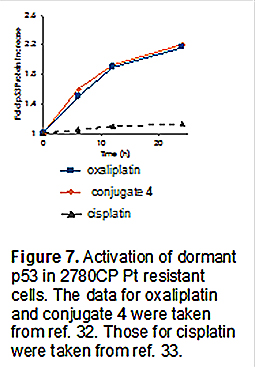
In accord with our design expectations, conjugate 4 was found to induce expression of p53 and its functional activation in both the A2780 and 2780CP cell lines, as evidenced by Western blotting and monitoring of the transcriptional activation of p21 as a downstream target of p53. Conjugate 4 was thus found to mirror the behavior seen for oxaliplatin, but not cisplatin, a platinum drug that is unable to induce p53 induction in the case of the resistant 2780CP cell line (Figure 7).
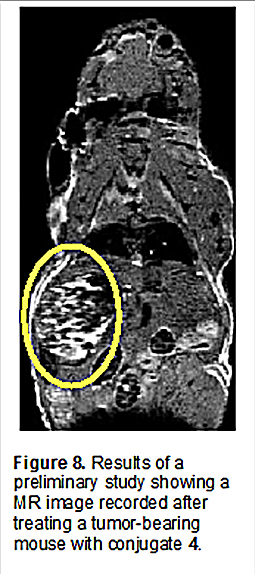
Unfortunately, efforts to translate the promise seen in the in vitro studies into xenograft murine tumor models proved challenging. Although in one preliminary study, evidence of tumor localization was seen through MR imaging (Figure 8), difficulties were encountered in finding a formulation adequate to allow for multiple tail vein injections.
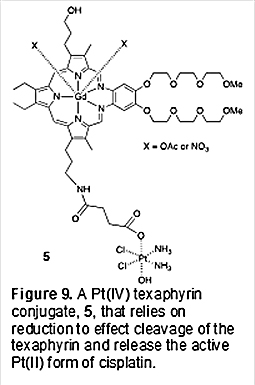
As a consequence, initial seemingly promising tumor uptake and growth inhibition results proved difficult to reproduce. A decision was thus made at this juncture to redesign our approach and 1) rely on Pt(IV) prodrug forms of FDA-approved platinum anticancer agents and 2) tether the Pt(IV) payload through the tripyrrane substituents, rather than the ortho-phenylenediamine subunit, as in the case of 3 and 4. This led to the production of second generation texaphyrin-platinum prodrug conjugates as detailed below. Second Generation Texaphyrin-platinum Prodrug Conjugates As yet, no Pt(IV) agent has received FDA approval as an anticancer drug. However, such species have been extensively explored as potential prodrugs [35]. The canonical thinking underlying these efforts is that the inherently reducing environment of solids tumors, a reflection of the Warburg effect, would lead to reduction of the Pt(IV) form to produce the corresponding Pt(II) form. Pt(IV) complexes typically favor an hexacoordinate octahedral coordination environment, while Pt(II) complexes are characterized by square planar tetracoordination. A consequence of this difference is that, with appropriate design, metal-centered reduction can be used to effect release of an active Pt(II) drug via loss of the axial ligands present in the Pt(IV) prodrug form. Pt(IV) species are generally exchange inert ; they are further attractive in that reduced off-target toxicity would be expected compared to the active Pt(II) species. This expected reduction in toxicity provided an incentive to prepare Pt(IV) texaphyrin conjugates [36]. Further motivation came from an appreciation that, relative to the corresponding Pt(II) species, Pt(IV) complexes are generally more water soluble, which was expected to aid in formulation. Given the above, we postulated that it would be possible to attach a gadolinium texaphyrin localizing core to a Pt(IV) prodrug form via a single axial carboxylate ligand. This ligation motif, in turn, would serve as a cleavable linker in the reduction that was expected to sever the resulting texaphyrin-platinum(II) connection. As a first test of this hypothesis, the Pt(IV) texaphyrin conjugate 5 (Figure 9) was prepared as reported by Thiabaud, et al. in 2014 [37].
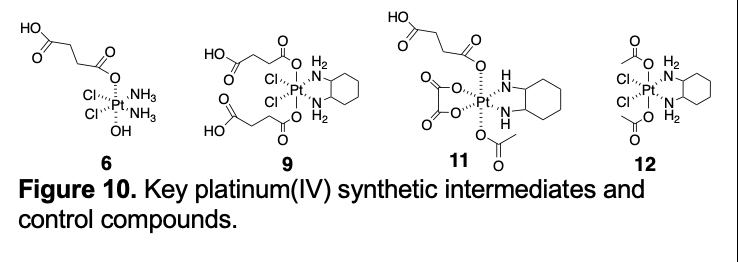
In this conjugate, the Pt(IV) center was designed to serve as a prodrug form of cisplatin. The critical design element is the axial carboxylate anion that serves to link the Pt(IV) center to the texaphyrin core. The requisite carboxylate species was put in place synthetically by coupling the succinyl functionalized Pt(IV) derivative, 6 (Figure 10), with the amine functionalized texaphryn 7.
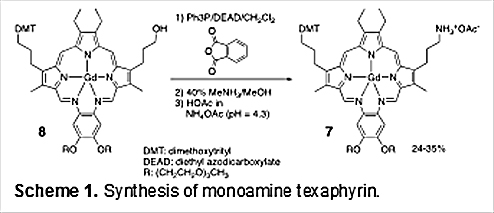
This key texaphyrin monoamine was prepared by subjecting MGd to mono-protection (to give 8), followed by Mitsunobu amination, as originally detailed by Magda and collaborators in 2004 (cf. Scheme 1) [20].
The synthesis of this key precursor remains the subject of ongoing optimization efforts in the author's laboratory. Conjugate 5 was found to be stable in the dark, but to undergo photo-induced reduction to release cisplatin under conditions of normal laboratory illumination. The increased hydrolytic stability relative to the earlier Pt(II) conjugates in the absence of light was considered to be an advantage, as was the apparent greater water solubility of 5 relative to, for example, conjugate 3. Next, in vitro studies, involving comparisons between the wild type and platinum resistant A2789 and 2780CP ovarian cancer cell lines in analogy to what was done for conjugates 3 and 4, were carried out. These studies revealed that conjugate 5 was somewhat more potent than 3 on a per platinum basis (IC50 = 1.3 mM vs 1.6 mM in the A2780 cell line). Unfortunately, and as expected for a cis-platinum prodrug, the resistance factor (defined as IC50(2780CP)/IC50(A2780)) was less than that seen for oxaliplatin; it was ca. 8 in the case of conjugate 5 (vs. ca. 2 in the case of oxaliplatin). Thus, a decision was made to create analogues of 5 that incorporated a Pt(IV) center that upon reductive cleavage would release oxaliplatin as the active drug form. A finding made subsequent to the publication of conjugates 5 and 6 provided a further incentive to prepare Pt(IV) prodrug forms of oxaliplatin. Specifically, on the basis of early studies showing that MGd would catalyze the reduction of dioxygen (to produce reactive oxygen species) in the presence of reducing metabolites that texaphyrins are not redox innocent [38], we came to appreciate that the gadolinium texaphyrin core might act as a redox cycler to facilitate the electron transfer-based conversion of Pt(IV) species to the corresponding Pt(II) forms. A study by Thiabaud, et al., reported in 2016, provided experimental support for this proposition. It was found, for instance, that an equimolar mixture of the bis-succinyl Pt(IV) analogue of oxaliplatin (compound 9; Figure 10) and MGd was more cytotoxic (by ca. 6x) than either species on its own [39].
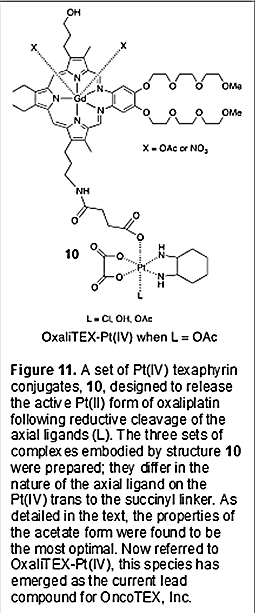
Given the above findings, an effort was made to prepare texaphyrin conjugates containing a Pt(IV) analogue of oxaliplatin. As detailed in a recent (2020) report by Thiabaud, et al., three mono-Pt(IV) derivatives (represented by generic structure 10; Figure 11) were thus prepared [40]. These systems differ only in the choice of the second axial ligand present on the Pt(IV) center. As in the case of conjugate 5, these complexes were generated by coupling the corresponding succinyl substituted precursor (e.g., 11 in the case of the acetate form of 10; Figure 10) with the monoamino texaphyrin derivative 7.
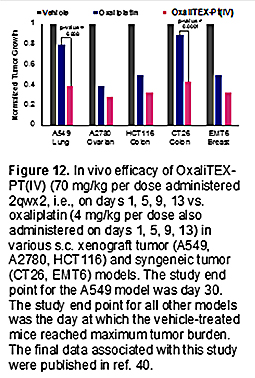
Since all three versions of conjugate 10 contain a Pt(IV) center, none was expected to be active on its own; all would require reductive cleavage to give oxaliplatin to exert an anticancer effect. They thus provided a test bed for exploring the effect of the non-conjugating axial ligand on the Pt(IV) center. In accord with the literature [41], the effect was found to be substantial. In the presence of glutathione, an endogenous reductant, the chloride ligated version was found to be 90% reduced (and thus cleaved) within 10 minutes upon incubation at 37 ºC. In contrast, both the acetate and hydroxyl ligated versions were relatively stable, remaining ca. 75% intact under these same incubation conditions for roughly 1 and 2 hours, respectively. This difference in relative stability was found to track with the redox potentials, with the chloride ligated species proving to be substantially easier to reduce than the other two forms. On the other hand, cell proliferation assays revealed that the acetate form of 10 was more potent by a factor of ca. 2 than the corresponding hydroxyl species. It was thus speculated that it represented a Goldilocks version of the complex wherein the inherent stability was high enough to allow for its use, while reduction remained sufficiently facile to allow for release of the active Pt(II) oxaliplatin form [40]. A doubly functionalized version of the acetate form of 10 (i.e., with both hydroxypropyl moieties of the tripyrrane replaced by the Pt(IV) oxaliplatin prodrug) was also prepared. However, its solubility features were less attractive than those of 10. In fact, the acetate version of conjugate 10, now referred to as OxaliTEX-Pt(IV), proved soluble in aqueous media. This feature made it very attractive in terms of formulation efforts. It was thus selected for in depth study [40]. Initial in vitro tests confirmed that, per our design expectations, there was no difference in the potency of OxaliTEX-Pt(IV) for the wild type and platinum resistance A2780 and 2780CP cell lines within error (resistance factor ≤1.2). OxaliTEX-Pt(IV) also proved more effective than a combination of MGd and the control Pt(IV) oxaliplatin analogue 12 (structure shown in Figure 10). OxaliTEX-Pt(IV) was further tested in a number of other cell lines, with potency being observed in all cases as inferred from MTT assays. In A549 human lung cancer cells, for instance, an IC50 of ca. 2.1 mM was recorded in an in vitro study involving a 5-day incubation period. Further, and as expected for an oxaliplatin prodrug, activation of p53 by OxaliTEX-Pt(IV) was seen in the platinum resistant 2780CP cells, as assessed by monitoring the transcriptional activation of p21 as a downstream target of p53. Finally, in vitro studies with the A549 cell line also revealed 1.7x more Pt being taken up into the cells as compared to oxaliplatin. This latter finding was taken as support for the hypothesis underlying the texaphyrin conjugate program, namely that the use of an MGd core aids in uptake into cancer cells. Based on the above, OxaliTEX-Pt(IV) was selected for study in vivo [40]. A number of animal studies were thus carried out using various mouse models. As a predicate to tests of efficacy, tolerability was tested using athymic nude mice. Repeat dose studies revealed that OxaliTEX-Pt(IV) was well tolerated with no significant body weight loss being observed over a 30-day period when mice were dosed via intravenous (i.v.) tail vein injection (60 mg/kg per dose) on days 1, 5, 9, and 13 (referred to as 2qwx2) with concurrent and subsequent monitoring. Confirmatory analyses revealed that up to 70 mg/kg could be tolerated. This is ca. 4x the limit found for oxaliplatin on an equivalent per-platinum (i.e., per mole) basis. Initial evidence of antitumor efficacy was seen in athymic nude mice bearing established subcutaneous (s.c.) A549 human lung cancer xenografts (n = 9 mice per group). At the study end point, day 28, mice treated with oxaliplatin exhibited statistically insignificant growth inhibition relative to the vehicle group. In contrast, statistically significant (relative to the oxaliplatin group) tumor growth inhibition was seen in xenograft-bearing mice treated i.v. with OxaliTEX-Pt(IV) (values of 51, 58, and 61%, respectively, for the 50, 60, and 70 mg/kg per dose cohorts). Efficacy studies involving OxaliTEX-Pt(IV) were also conducted in mice bearing cell-derived (A2780 ovarian and HCT116 colon) and syngeneic tumors (CT26 colon and EMT6 breast) s.c. xenografts. Statistically significant anticancer activity against all four tumor types was seen relative to both the vehicle and oxaliplatin-treated mice. In these studies, OxaliTex-Pt(IV) and oxaliplatin were studied near their respective maximum tolerated doses (MTDs) of 70 mg/kg and 4 mg/kg, with administration being effected via tail vein injection on days 1, 5, 9, and 13 in both cases (Figure 12).
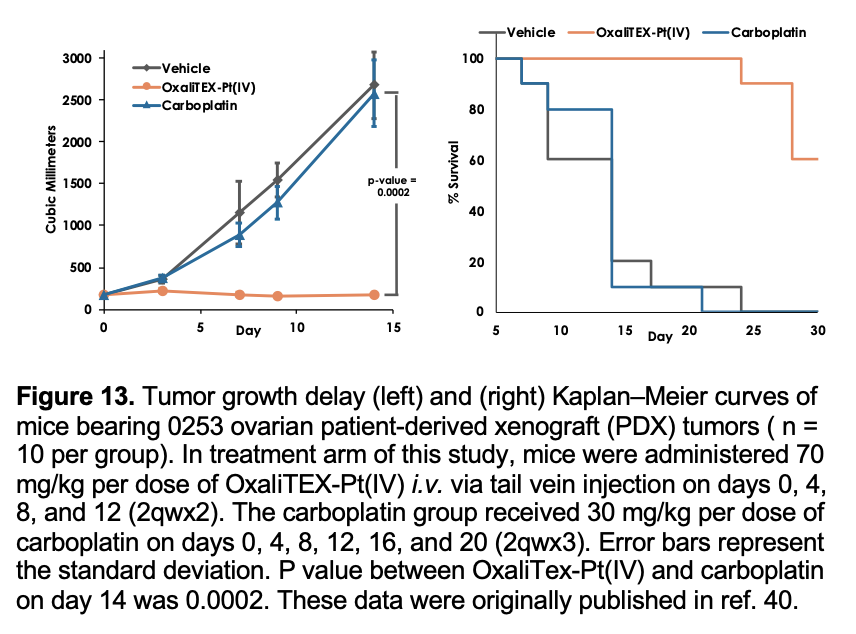
To assess whether OxaliTEX-Pt(IV) might have a role to play in treating tumors that are characterized by poor responses to traditional platinum agents, further efficacy studies in patient-derived xenografts (PDXs), including the 0253 ovarian cancer xenograft (provided by Champions Oncology), were carried out. Mice bearing 0253 ovarian PDX tumors did not respond to treatment with carboplatin (the current standard of care platinum drug for this disease) when administered at a dose level near its MTD. In contrast, treatment with OxaliTEX-Pt(IV), again at a dose close to the MTD, resulted in 95% tumor growth inhibition at the end of the study (Figure 13, left). This delay also translated to a statistically significant increase in survival (Figure 13, right).
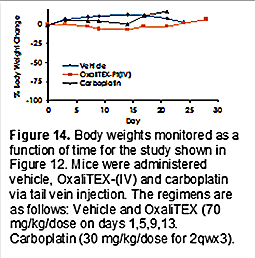
Promising results were also found in related studies involving the 0069 colorectal PDX model. In neither case was appreciable weight loss seen during the course of the studies (cf., e.g., Figure 14). This latter finding was taken as evidence that, at the near-MTD doses administered, OxaliTEX-Pt(IV) was well tolerated.
As a complement to the efficacy studies, initial biodistribution studies were carried out. It was found that the conjugation of the Pt(IV) prodrug that makes up OxaliTEX-Pt(IV) to the amphiphilic gadolinium texaphyrin core, serves to alter the biodistribution away from the kidney and more toward the liver relative to what is normally seen for platinum drugs. This redistribution and presumed change in dominant clearance pathways may prove beneficial in a clinical setting given the renal toxicity issues that have long plagued cisplatin. In a separate experiment involving mice bearing HCT116 colon xenografts, a statistically significant increased level of tumor tissue localization was seen for OxaliTEX-Pt(IV) relative to oxaliplatin (based on analyses of excised tumors) after normalization to the amount of agent administered. Localization of OxaliTEX-Pt(IV) in cancerous lesions in preference to healthy tissues was further corroborated with pharmacokinitec studies in mice. It was found that that the low-level exposure to "free" Pt contributes to drug tolerance in the case of normal tissues, whereas tumor-targeting as provided by OxaliTEX, contributes to greater tumor uptake that enhances antitumor effects [42]. This was taken as further support for the notion that conjugation of an active drug or prodrug to a texaphyrin core can lead to enhancements in tumor localization. Thus, this result serves not just to underscore the promise inherent in OxaliTEX-Pt(IV) as a drug lead, it also validates ongoing efforts to develop texaphyrins as tumor-targeting carriers for a range of active therapeutics beyond platinum.Conclusion In September of 2019 the intellectual property rights [43,44] underpinning the texaphyrin drug conjugate program were licensed to the IQ Global Group by The University of Texas at Austin and MD Anderson Cancer Center [45]. Active efforts are now ongoing to streamline and scaleup the synthesis of OxaliTEX-Pt(IV), including so-called GMP manufacturing, and carry out IND-enabling preclinical studies involving a variety of additional animal models. In parallel, work is being devoted to the creation of new texaphyrin conjugates wherein both the central texaphyrin core and the nature of the appended active agent are varied. It is expected that the results of these efforts will be communicated in due course. Acknowledgments The author expresses his gratitude to the many team members who have helped develop the texaphyrin-platinum conjugate project, including Darren J. Magda, Richard A. Miller, Tarak Mody, Pavel Anzenbacher, Jr., Joan Carvalho, Mark Fountain, Wen-hao Wei, Jonathan F. Arambula, Grégory Thiabaud, Kwan Soo Hong, Greg Lyness, Alan B. Watts, Zahid Siddik, Guangan He, Rick Finch, Sajal Sen, Calvin Chao, and Adam Sedgwick. Over the course of the project lifetime, funding for the work in Austin has been provided by the National Institutes of Health (grant CA68682), Pharmacyclics, Inc., The University of Texas at Austin TI-3D (supported by the Robert A. Welch Foundation, grant H-F-0032), the American Cancer Society (grant PF-11-015-01-CDD to J. F. Arambula) the Cancer Prevention and Research Institute of Texas (grants RP120393 and RP140108). Current project funding is provided by OncoTEX, and the Robert A. Welch Foundation (F-1018 References
|
||||||||||||||||||||||||||||||||||||||||||||||||